- British Pharmacopoeia Volume I & II
- Monographs: Medicinal and Pharmaceutical Substances
Microcrystalline Cellulose |
 |
(Ph. Eur. monograph 0316)
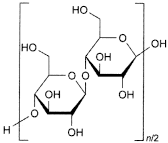
C6nH10n+2O5n+1 9004-34-6
Excipient.
Ph Eur
Purified, partly depolymerised cellulose prepared by treating alpha-cellulose, obtained as a pulp from fibrous plant material, with mineral acids.
White or almost white, fine or granular powder.
Practically insoluble in water, in acetone, in anhydrous ethanol, in toluene, in dilute acids and in a 50 g/L solution of sodium hydroxide.
A. Place about 10 mg on a watch-glass and disperse in 2 mL of iodinated zinc chloride solution R. The substance becomes violet-blue.
B. The degree of polymerisation is not more than 350.
Transfer 1.300 g to a 125 mL conical flask. Add 25.0 mL of water R and 25.0 mL of cupriethylenediamine hydroxide solution R. Immediately purge the solution with nitrogen R, insert the stopper and shake until completely dissolved. Transfer an appropriate volume of the solution to a suitable capillary viscometer (2.2.9). Equilibrate the solution at 25 ± 0.1 °C for at least 5 min. Record the flow time (t1) in seconds between the 2 marks on the viscometer. Calculate the kinematic viscosity (ν1) of the solution using the following expression:
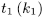
where k1 is the viscometer constant.
Dilute a suitable volume of cupriethylenediamine hydroxide solution R with an equal volume of water R and measure the flow time (t2) using a suitable capillary viscometer. Calculate the kinematic viscosity (ν2) of the solvent using the following expression:
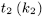
where k2 is the viscometer constant.
Determine the relative viscosity (ηrel) of the substance to be examined using the following expression:
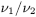
Determine the intrinsic viscosity ([η]c) by interpolation, using the intrinsic viscosity table (Table 0316.-1).
Calculate the degree of polymerisation (P) using the following expression:
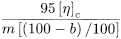
where m is the mass in grams of the substance to be examined and b is the loss on drying as a percentage.
Dissolve 50 mg in 10 mL of ammoniacal solution of copper tetrammine R. It dissolves completely, leaving no residue.
5.0 to 7.5 for the supernatant liquid.
Shake 5 g with 40 mL of carbon dioxide-free water R for 20 min and centrifuge.
The conductivity of the test solution does not exceed the conductivity of the water by more than 75 µS·cm-1.
Use as test solution the supernatant liquid obtained in the test for pH. Measure the conductivity of the supernatant liquid after a stable reading has been obtained and measure the conductivity of the water used to prepare the test solution.
Maximum 0.05 per cent (5 mg) for the difference between the weight of the residue and the weight obtained from a blank determination.
Place 10.0 g in a chromatography column about 20 mm in internal diameter and pass 50 mL of peroxide-free ether R through the column. Evaporate the eluate to dryness. Dry the residue at 105 °C for 30 min, allow to cool in a desiccator and weigh. Carry out a blank determination.
Maximum 0.25 per cent (12.5 mg) for the difference between the mass of the residue and the mass obtained from a blank determination.
Shake 5.0 g with 80 mL of water R for 10 min. Filter through a filter paper with the aid of vacuum into a tared flask. Evaporate to dryness on a water-bath avoiding charring. Dry at 105 °C for 1 h, allow to stand in a desiccator and weigh. Carry out a blank determination.
Maximum 10 ppm.
2.0 g complies with test C. Prepare the reference solution using 2 mL of lead standard solution (10 ppm Pb) R.
Maximum 7.0 per cent, determined on 1.000 g by drying in an oven at 105 °C for 3 h.
Maximum 0.1 per cent, determined on 1.0 g.
TAMC: acceptance criterion 103 CFU/g (2.6.12).
TYMC: acceptance criterion 102 CFU/g (2.6.12).
Absence of Escherichia coli (2.6.13).
Absence of Pseudomonas aeruginosa (2.6.13).
Absence of Staphylococcus aureus (2.6.13).
Absence of Salmonella (2.6.13).
This section provides information on characteristics that are recognised as being relevant control parameters for one or more functions of the substance when used as an excipient (see chapter 5.15). This section is a non-mandatory part of the monograph and it is not necessary to verify the characteristics to demonstrate compliance. Control of these characteristics can however contribute to the quality of a medicinal product by improving the consistency of the manufacturing process and the performance of the medicinal product during use. Where control methods are cited, they are recognised as being suitable for the purpose, but other methods can also be used. Wherever results for a particular characteristic are reported, the control method must be indicated.The following characteristics may be relevant for microcrystalline cellulose used as binder, diluent or disintegrant.



Ph Eur