- British Pharmacopoeia Volume I & II
- Monographs: Medicinal and Pharmaceutical Substances
Tioconazole |
 |


(Ph. Eur. monograph 2074)
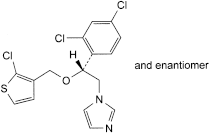
C16H13Cl3N2OS 387.7 65899-73-2
Antifungal
Ph Eur
1-[(2RS)-2-[(2-Chlorothiophen-3-yl)methoxy]-2-(2,4-dichlorophenyl)ethyl]-1H-imidazole.
99.0 per cent to 101.0 per cent (anhydrous substance).
White or almost white, crystalline powder.
Very slightly soluble in water, very soluble in methylene chloride, freely soluble in alcohol.
Infrared absorption spectrophotometry (2.2.24).
Comparison Ph. Eur. reference spectrum of tioconazole.
Liquid chromatography (2.2.29).
Test solution Dissolve 20.0 mg of the substance to be examined in the mobile phase and dilute to 10.0 mL with the mobile phase.
Reference solution (a) Dilute 1.0 mL of the test solution to 100.0 mL with the mobile phase. Dilute 2.0 mL of this solution to 10.0 mL with the mobile phase.
Reference solution (b) Dissolve 5 mg of tioconazole for system suitability CRS in the mobile phase and dilute to 2.5 mL with the mobile phase.
- — size: l = 0.25 m, Ø = 4.6 mm,
- — stationary phase: end-capped octadecylsilyl silica gel for chromatography R (5 µm) with a specific surface area of 170 m2/g, a pore size of 12 nm and a carbon loading of 10 per cent.
Mobile phase Mix 1 volume of a 1.7 g/L solution of tetrabutylammonium dihydrogen phosphate R previously adjusted to pH 7.4 with dilute ammonia R2 and 3 volumes of methanol R.
Flow rate 1 mL/min.
Detection Spectrophotometer at 218 nm.
Injection 20 µL.
Run time 2.5 times the retention time of tioconazole.
System suitability Reference solution (b):
- — resolution: minimum 1.0 between the peaks due to impurity B and impurity C (locate impurities A, B and C by comparison with the chromatogram provided with tioconazole for system suitability CRS).
- — correction factors: for the calculation of contents, multiply the peak areas of the following impurities by the corresponding correction factor: impurity B = 1.7; impurity C = 1.7.
- — impurities A, B, C: for each impurity, not more than 1.5 times the area of the principal peak in the chromatogram obtained with reference solution (a) (0.3 per cent),
- — unspecified impurities: for each impurity, not more than 0.5 times the area of the principal peak in the chromatogram obtained with reference solution (a) (0.10 per cent),
- — total: not more than 5 times the area of the principal peak in the chromatogram obtained with reference solution (a) (1.0 per cent),
- — disregard limit: 0.25 times the area of the principal peak in the chromatogram obtained with reference solution (a) (0.05 per cent).
Maximum 0.5 per cent, determined on 1.00 g.
Maximum 0.1 per cent, determined on 1.0 g.
Dissolve 0.300 g in 50 mL of anhydrous acetic acid R. Titrate with 0.1 M perchloric acid, determining the end-point potentiometrically (2.2.20).
1 mL of 0.1 M perchloric acid is equivalent to 38.77 mg of C16H13Cl3N2OS.
Protected from light.
Specified impurities A, B, C.
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10. Control of impurities in substances for pharmaceutical use): D.
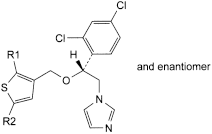
A. R1 = R2 = H: 1-[(2RS)-2-(2,4-dichlorophenyl)-2-[(thiophen-3-yl)methoxy]ethyl]-1H-imidazole,
B. R1 = R2 = Cl: 1-[(2RS)-2-(2,4-dichlorophenyl)-2-[(2,5-dichlorothiophen-3-yl)methoxy]ethyl]-1H-imidazole,
C. R1 = Cl, R2 = Br: 1-[(2RS)-2-[(5-bromo-2-chlorothiophen-3-yl)methoxy]-2-(2,4-dichlorophenyl)ethyl]-1H-imidazole,
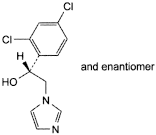
D. (1RS)-1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethanol.
Ph Eur