- British Pharmacopoeia Volume IV
- Appendices
Appendix III Chromatographic Separation Techniques |

Chromatographic separation techniques are multi-stage separation methods in which the components of a sample are distributed between 2 phases, one of which is stationary, while the other is mobile. The stationary phase may be a solid or a liquid supported on a solid or a gel. The stationary phase may be packed in a column, spread as a layer, or distributed as a film, etc. The mobile phase may be gaseous or liquid or supercritical fluid. The separation may be based on adsorption, mass distribution (partition), ion exchange, etc., or may be based on differences in the physico-chemical properties of the molecules such as size, mass, volume, etc.
This chapter contains definitions and calculations of common parameters and generally applicable requirements for system suitability. Principles of separation, apparatus and methods are given in the following general methods:
The system suitability and acceptance criteria in monographs have been set using parameters as defined below. With some equipment, certain parameters, such as the signal-to-noise ratio and resolution, can be calculated using software provided by the manufacturer. It is the responsibility of the user to ensure that the calculation methods used in the software are equivalent to the requirements of the European Pharmacopoeia and to make any necessary corrections if this is not the case.
A graphical or other representation of detector response, effluent concentration or other quantity used as a measure of effluent concentration, versus time or volume. Idealised chromatograms are represented as a sequence of Gaussian peaks on a baseline (Figure 2.2.46.-1).

The portion of a chromatogram recording the detector response when a single component (or 2 or more unresolved components) is eluted from the column.
The peak may be defined by the peak area, or the peak height (h) and the peak width at half-height (w h ), or the peak height (h) and the peak width between the points of inflection (w i ). In Gaussian peaks (Figure 2.2.46.-1) there is the following relationship:

Time required for elution of a component (Figure 2.2.46.-1, baseline scale being in minutes).
Volume of the mobile phase required for elution of a component. It may be calculated from the retention time and the flow rate (F) in millilitres per minute using the following equation:

Time required for elution of an unretained component (Figure 2.2.46.-1, baseline scale being in minutes). In size-exclusion chromatography, the symbol t 0 (see below) is used.
Volume of the mobile phase required for elution of an unretained component. It may be calculated from the hold-up time and the flow rate (F) in millilitres per minute using the following equation:
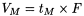
In size-exclusion chromatography, the symbol V 0 (see below) is used.

The retention factor (also known as mass distribution ratio (D m ) or capacity factor (k ′)) is defined as:

K C |
= |
distribution constant (also known as equilibrium distribution coefficient); |
V S |
= |
volume of the stationary phase; |
V M |
= |
volume of the mobile phase. |
The retention factor of a component may be determined from the chromatogram using the following equation:
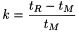
In size-exclusion chromatography, retention time of a component whose molecules are smaller than the smallest gel pores (Figure 2.2.46.-2).
In size-exclusion chromatography, retention volume of a component whose molecules are smaller than the smallest gel pores. It may be calculated from the total mobile phase time and the flow rate (F) in millilitres per minute using the following equation:
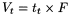
In size-exclusion chromatography, retention time of a component whose molecules are larger than the largest gel pores (Figure 2.2.46.-2).
In size-exclusion chromatography, retention volume of a component whose molecules are larger than the largest gel pores. It may be calculated from the retention time of an unretained compound and the flow rate (F) in millilitres per minute using the following equation:
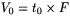
In size-exclusion chromatography, the elution characteristics of a component in a particular column may be given by the distribution constant (also referred to as distribution coefficient), which is calculated using the following equation:

The retardation factor (also known as retention factor (R f )), used in planar chromatography, is the ratio of the distance from the point of application to the centre of the spot and the distance travelled by the solvent front from the point of application (Figure 2.2.46.-3).

b |
= |
migration distance of the component; |
a |
= |
migration distance of the solvent front. |

The column performance (apparent efficiency) may be calculated from data obtained under either isothermal, isocratic or isodense conditions, depending on the technique, as the plate number (also referred to as number of theoretical plates), using the following equation, the values of t R and w h being expressed in the same units:
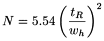
t R |
= |
retention time of the peak corresponding to the component; |
w h |
= |
width of the peak at half-height. |
The plate number varies with the component as well as with the column, the column temperature, the mobile phase and the retention time.
The dwell volume (also known as gradient delay volume) is the volume between the point at which the eluents meet and the top of the column. It can be determined using the following procedure.
Column replace the chromatographic column by an appropriate capillary tubing (e.g. 1 m × 0.12 mm).
Mobile phase:
- — mobile phase A: water R ;
- — mobile phase B: 0.1 per cent V/V solution of acetone R ;

Flow rate set to obtain sufficient back-pressure (e.g. 2 ml/min).
Detection spectrophotometer at 265 nm.
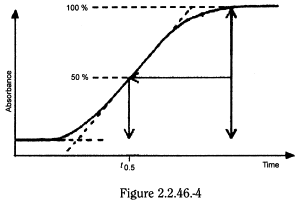
Determine the time (t 0.5) in minutes when the absorbance has increased by 50 per cent (Figure 2.2.46.-4).
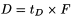
t D |
= |
t 0.5 - 0.5t G (in minutes); |
t G |
= |
pre-defined gradient time (= 20 min); |
F |
= |
flow rate (in millilitres per minute). |
The symmetry factor of a peak (Figure 2.2.46.-5) is calculated using the following equation:

w 0.05 |
= |
width of the peak at one-twentieth of the peak height; |
d |
= |
distance between the perpendicular dropped from the peak maximum and the leading edge of the peak at one-twentieth of the peak height. |
An A s value of 1.0 signifies symmetry. When A s > 1.0, the peak is tailing. When A s << 1.0, the peak is fronting.
The resolution between peaks of 2 components (Figure 2.2.46.-1) may be calculated using the following equation:
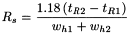
t R 2 >> t R 1 |
||
t R 1, t R 2 |
= |
retention times of the peaks; |
w h 1, w h 2 |
= |
peak widths at half-height. |

In quantitative planar chromatography, using densitometry, the migration distances are used instead of retention times and the resolution between peaks of 2 components may be calculated using the following equation:

R F 1, R F 2 |
= |
retardation factors of the peaks; |
w h 1, w h 2 |
= |
peak widths at half-height; |
a |
= |
migration distance of the solvent front. |
The peak-to-valley ratio may be employed as a system suitability criterion in a test for related substances when baseline separation between 2 peaks is not achieved (Figure 2.2.46.-6).
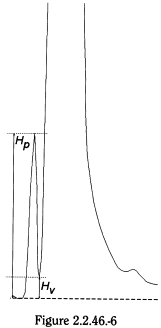

H p |
= |
height above the extrapolated baseline of the minor peak; |
H v |
= |
height above the extrapolated baseline at the lowest point of the curve separating the minor and major peaks. |
Relative retention is calculated as an estimate using the following equation:

t Ri |
= |
retention time of the peak of interest; |
t Rst |
= |
retention time of the reference peak (usually the peak corresponding to the substance to be examined); |
t M |
= |
hold-up time. |
The unadjusted relative retention (r G ) is calculated using the following equation:

Unless otherwise indicated, values for relative retention stated in monographs correspond to unadjusted relative retention.
In planar chromatography, the retardation factors R Fst and R Fi are used instead of t Rst and t Ri .
The short-term noise influences the precision of quantification. The signal-to-noise ratio is calculated using the following equation:

H |
= |
height of the peak (Figure 2.2.46.-7) corresponding to the component concerned, in the chromatogram obtained with the prescribed reference solution, measured from the maximum of the peak to the extrapolated baseline of the signal observed over a distance equal to at least 5 times the width at half-height; |
h |
= |
range of the noise in a chromatogram obtained after injection or application of a blank, observed over a distance equal to at least 5 times the width at half-height of the peak in the chromatogram obtained with the prescribed reference solution and, if possible, situated equally around the place where this peak would be found. |

The repeatability of response is expressed as an estimated percentage relative standard deviation (s r (%)) of a consecutive series of measurements for not fewer than 3 injections or applications of a reference solution, and is calculated using the following equation:

y i |
= |
individual values expressed as peak area, peak height, or ratio of areas by the internal standardisation method; |
|
= |
mean of individual values; |
n |
= |
number of individual values. |
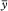
The various components of the equipment employed must be qualified and be capable of achieving the performance required to conduct the test or assay.
The system suitability tests represent an integral part of the method and are used to ensure adequate performance of the chromatographic system. Apparent efficiency, retention factor (mass distribution ratio), resolution, relative retention and symmetry factor are the parameters that are usually employed in assessing the performance of the column. Factors that may affect the chromatographic behaviour include:
- — the composition, ionic strength, temperature and apparent pH of the mobile phase;
- — flow rate, column dimensions, column temperature and pressure;
- — stationary phase characteristics including type of chromatographic support (particle-based or monolithic), particle or macropore size, porosity, specific surface area;
- — reversed-phase and other surface-modification of the stationary phases, the extent of chemical modification (as expressed by end-capping, carbon loading etc.).
The following requirements and any supplementary requirements given in the individual monograph are to be fulfilled unless otherwise prescribed:
- — in a related substances test or assay, for a peak in the chromatogram obtained with a reference solution used for quantification, the symmetry factor is 0.8 to 1.5, unless otherwise prescribed;
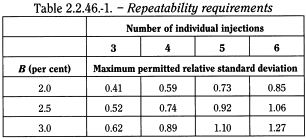
- — in an assay of an active substance where the value is 100 per cent for a pure substance, the maximum permitted relative standard deviation (s r (%)max) for the defined limits is calculated for a series of injections of the reference solution using the following equation:

K |
= |
constant (0.349), obtained from the expression |
B |
= |
upper limit given in the definition of the individual monograph minus 100 per cent; |
n |
= |
number of replicate injections of the reference solution (3 ≤ n ≤ 6); |
t 90%, n - 1 |
= |
Student's t at the 90 per cent probability level (double sided) with n-1 degrees of freedom. |
 in which
in which  represents the required percentage relative standard deviation after 6 injections for
represents the required percentage relative standard deviation after 6 injections for Unless otherwise prescribed, the maximum permitted relative standard deviation does not exceed the appropriate value given in Table 2.2.46.-1. This requirement does not apply to tests for related substances.
- — in a related substances test, the limit of quantification (corresponding to a signal-to-noise ratio of 10) is equal to or less than the disregard limit.
Compliance with the system suitability criteria is required throughout the chromatographic procedure. Depending on various factors, such as the frequency of use of the procedure and experience with the chromatographic system, the analyst chooses an appropriate verification scheme to monitor this.
The extent to which the various parameters of a chromatographic test may be adjusted to satisfy the system suitability criteria without fundamentally modifying the methods are listed below. Adjustment of conditions with gradient elutions is more critical than with isocratic elutions, since it may lead to shifts in peaks to a different step of the gradient, thus leading to the incorrect assignment of peaks, and to the masking of peaks or a shift such that elution occurs beyond the prescribed elution time. Changes other than those indicated require revalidation of the method. The chromatographic conditions described have been validated during the elaboration of the monograph.
The system suitability tests are included to verify that the separation required for satisfactory performance of the test or assay is achieved. Nonetheless, since the stationary phases are described in a general way and there is such a variety available commercially, with differences in chromatographic behaviour, some adjustments of the chromatographic conditions may be necessary to achieve the prescribed system suitability requirements. With reversed-phase liquid chromatographic methods in particular, adjustment of the various parameters will not always result in satisfactory chromatography. In that case, it may be necessary to replace the column with another of the same type (e.g. octadecylsilyl silica gel), which exhibits the desired chromatographic behaviour. The Knowledge database on the EDQM website usually contains information on the column(s) used during monograph elaboration.
For critical parameters the adjustments are defined clearly in the monograph to ensure the system suitability.
Composition of the mobile phase the amount of the minor solvent component may be adjusted by ± 30 per cent relative or ± 2 per cent absolute, whichever is the larger; for a minor component at 10 per cent of the mobile phase, a 30 per cent relative adjustment allows a range of 7-13 per cent whereas a 2 per cent absolute adjustment allows a range of 8-12 per cent, the relative value therefore being the larger; for a minor component at 5 per cent of the mobile phase, a 30 per cent relative adjustment allows a range of 3.5-6.5 per cent whereas a 2 per cent absolute adjustment allows a range of 3-7 per cent, the absolute value being the larger in this case; no other component is altered by more than 10 per cent absolute.
pH of the aqueous component of the mobile phase ± 0.2 pH, unless otherwise prescribed, or ± 1.0 pH when non-ionisable substances are to be examined.
Concentration of salts in the buffer component of a mobile phase ± 10 per cent.
Application volume 10-20 per cent of the prescribed volume if using fine particle size plates (2-10 µm).
Composition of the mobile phase the amount of the minor solvent component may be adjusted by ± 30 per cent relative or ± 2 per cent absolute, whichever is the larger (see example above); no other component is altered by more than 10 per cent absolute.
pH of the aqueous component of the mobile phase ± 0.2 pH, unless otherwise prescribed, or ± 1.0 pH when non-ionisable substances are to be examined.
Concentration of salts in the buffer component of a mobile phase ± 10 per cent.
Flow rate ± 50 per cent; a larger adjustment is acceptable when changing the column dimensions (see the formula below).
Column parameters
Stationary phase:
- — no change of the identity of the substituent of the stationary phase permitted (e.g. no replacement of C18 by C8);
- — particle size: maximum reduction of 50 per cent; no increase permitted.
Column dimensions:
- — length: ± 70 per cent;
- — internal diameter: ± 25 per cent.
When column dimensions are changed, the flow rate may be adjusted as necessary using the following equation:
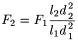
F 1 |
= |
flow rate indicated in the monograph, in millilitres per minute; |
F 2 |
= |
adjusted flow rate, in millilitres per minute; |
l 1 |
= |
length of the column indicated in the monograph, in millimetres; |
l 2 |
= |
length of the column used, in millimetres; |
d 1 |
= |
internal diameter of the column indicated in the monograph, in millimetres; |
d 2 |
= |
internal diameter of the column used, in millimetres. |
Temperature ± 10 °C, where the operating temperature is specified, unless otherwise prescribed.
Detector wavelength no adjustment permitted.
Injection volume may be decreased, provided detection and repeatability of the peak(s) to be determined are satisfactory; no increase permitted.
Adjustment of chromatographic conditions for gradient systems requires greater caution than for isocratic systems.
Composition of the mobile phase/gradient elution minor adjustments of the composition of the mobile phase and the gradient are acceptable provided that:
- — the system suitability requirements are fulfilled;
- — the principal peak(s) elute(s) within ± 15 per cent of the indicated retention time(s);
- — the final composition of the mobile phase is not weaker in elution power than the prescribed composition.
Where compliance with the system suitability requirements cannot be achieved, it is often preferable to consider the dwell volume or to change the column.
Dwell volume The configuration of the equipment employed may significantly alter the resolution, retention time and relative retentions described. Should this occur, it may be due to excessive dwell volume. Monographs preferably include an isocratic step before the start of the gradient programme so that an adaptation can be made to the gradient time points to take account of differences in dwell volume between the system used for method development and that actually used. It is the user's responsibility to adapt the length of the isocratic step to the analytical equipment used. If the dwell volume used during the elaboration of the monograph is given in the monograph, the time points (t min) stated in the gradient table may be replaced by adapted time points (t c min), calculated using the following equation:
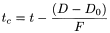
D |
= |
dwell volume, in millilitres; |
D 0 |
= |
dwell volume used for development of the method, in millilitres; |
F |
= |
flow rate, in millilitres per minute. |
The isocratic step introduced for this purpose may be omitted if validation data for application of the method without this step is available.
pH of the aqueous component of the mobile phase no adjustment permitted.
Concentration of salts in the buffer component of a mobile phase no adjustment permitted.
Flow rate adjustment is acceptable when changing the column dimensions (see the formula below).
Column parameters
Stationary phase:
- — no change of the identity of the substituent of the stationary phase permitted (e.g. no replacement of C18 by C8);
- — particle size: no adjustment permitted.
Column dimensions:
- — length: ± 70 per cent;
- — internal diameter: ± 25 per cent.
When column dimensions are changed, the flow rate may be adjusted as necessary using the following equation:

F 1 |
= |
flow rate indicated in the monograph, in millilitres per minute; |
F 2 |
= |
adjusted flow rate, in millilitres per minute; |
l 1 |
= |
length of the column indicated in the monograph, in millimetres; |
l 2 |
= |
length of the column used, in millimetres; |
d 1 |
= |
internal diameter of the column indicated in the monograph, in millimetres; |
d 2 |
= |
internal diameter of the column used, in millimetres. |
Temperature ± 5 °C, where the operating temperature is specified, unless otherwise prescribed.
Detector wavelength no adjustment permitted.
Injection volume may be decreased, provided detection and repeatability of the peak(s) to be determined are satisfactory; no increase permitted.
Column parameters
Stationary phase:
- — particle size: maximum reduction of 50 per cent; no increase permitted (packed columns);
- — film thickness: - 50 per cent to + 100 per cent (capillary columns).
Column dimensions:
- — length: ± 70 per cent;
- — internal diameter: ± 50 per cent.
Flow rate ± 50 per cent.
Temperature ± 10 per cent.
Injection volume and split volume may be adjusted, provided detection and repeatability are satisfactory.
Composition of the mobile phase for packed columns, the amount of the minor solvent component may be adjusted by ± 30 per cent relative or ± 2 per cent absolute, whichever is the larger; no adjustment is permitted for a capillary column system.
Detector wavelength no adjustment permitted.
Column parameters
Stationary phase:
- — particle size: maximum reduction of 50 per cent; no increase permitted (packed columns).
Column dimensions:
- — length: ± 70 per cent;
- — internal diameter:
± 25 per cent (packed columns);
± 50 per cent (capillary columns).
Flow rate ± 50 per cent.
Temperature ± 5 °C, where the operating temperature is specified.
Injection volume may be decreased, provided detection and repeatability are satisfactory; no increase permitted.
Peaks due to solvents and reagents or arising from the mobile phase or the sample matrix are disregarded during quantification.
- — Detector sensitivity. The detector sensitivity is the signal output per unit concentration or unit mass of a substance in the mobile phase entering the detector. The relative detector response factor, commonly referred to as response factor, expresses the sensitivity of a detector for a given substance relative to a standard substance. The correction factor is the reciprocal of the response factor.
- — External standard method. The concentration of the component(s) to be analysed is determined by comparing the response(s) (peak(s)) obtained with the test solution to the response(s) (peak(s)) obtained with a reference solution.
- — Internal standard method. Equal amounts of a component that will be resolved from the substance to be examined (the internal standard) are introduced into the test solution and a reference solution. The internal standard is chosen such that it does not react with the substance to be examined, is stable and does not contain impurities with the same retention time as that of the substance to be examined. The concentration of the substance to be examined is determined by comparing the ratio of the peak areas or peak heights due to the substance to be examined and the internal standard in the test solution with the ratio of the peak areas or peak heights due to the substance to be examined and the internal standard in the reference solution.
- — Normalisation procedure . The percentage content of a component of the substance to be examined is calculated by determining the area of the corresponding peak as a percentage of the total area of all the peaks, excluding those due to solvents or reagents or arising from the mobile phase or the sample matrix, and those at or below the disregard limit.
- — Calibration procedure. The relationship between the measured or evaluated signal (y) and the quantity (concentration, mass, etc.) of substance (x) is determined and the calibration function is calculated. The analytical results are calculated from the measured signal or evaluated signal of the analyte by means of the inverse function.
In tests for related substances for both the external standard method, when a dilution of the test solution is used for comparison, and the normalisation procedure, any correction factors indicated in the monograph are applied (i.e. when the response factor is outside the range 0.8-1.2).
When the related substances test prescribes the total of impurities or there is a quantitative determination of an impurity, it is important to choose an appropriate threshold setting and appropriate conditions for the integration of the peak areas. In such tests the disregard limit, i.e. the limit at or below which a peak is disregarded, is generally 0.05 per cent. Thus, the threshold setting of the data collection system corresponds to at least half of the disregard limit. Integration of the peak area of any impurity that is not completely separated from the principal peak is preferably performed by valley-to-valley extrapolation (tangential skim).
Unless otherwise stated in the monograph, the maximum permitted relative standard deviation for six replicate injections of the prescribed reference solution does not exceed 2.0%. This requirement is applicable only to assays.