- British Pharmacopoeia Volume I & II
- Monographs: Medicinal and Pharmaceutical Substances
Valsartan |
 |
(Ph. Eur. monograph 2423)
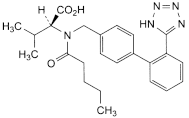
C24H29N5O3 435.5 137862-53-4
Angiotensis II (AT1) receptor antagonist.
Ph Eur
(2S)-3-Methyl-2-[pentanoyl[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]amino]butanoic acid.
99.0 per cent to 101.0 per cent (anhydrous substance).
White or almost white, hygroscopic powder.
Practically insoluble in water, freely soluble in anhydrous ethanol, sparingly soluble in methylene chloride.
Carry out either tests A, B or tests A, C.
A. Infrared absorption spectrophotometry (2.2.24).
Comparison valsartan CRS.
B. Enantiomeric purity (see Tests).
C. Specific optical rotation (2.2.7): - 64.0 to - 69.0 (anhydrous substance).
Dissolve 0.200 g in methanol R and dilute to 20.0 mL with the same solvent.
Liquid chromatography (2.2.29).
Test solution Dissolve 50 mg of the substance to be examined in the mobile phase and dilute to 50.0 mL with the mobile phase.
Reference solution (a) Dissolve 5 mg of valsartan for peak identification CRS (containing impurity A) in the mobile phase and dilute to 5.0 mL with the mobile phase.
Reference solution (b) Dilute 1.0 mL of the test solution to 100.0 mL with the mobile phase.
- — size: l = 0.25 m, Ø = 4.6 mm;
- — stationary phase: silica gel OD for chiral separations R.
Mobile phase trifluoroacetic acid R, 2-propanol R, hexane R (0.1:15:85 V/V/V).
Flow rate 0.8 mL/min.
Detection Spectrophotometer at 230 nm.
Injection 10 µL.
Run time 1.5 times the retention time of valsartan.
Identification of impurities Use the chromatogram supplied with valsartan for peak identification CRS and the chromatogram obtained with reference solution (a) to identify the peak due to impurity A.
Relative retention With reference to valsartan (retention time = about 13 min): impurity A = about 0.6.
System suitability Reference solution (a):
- — resolution: minimum 2.0 between the peaks due to impurity A and valsartan.
- — impurity A: not more than the area of the principal peak in the chromatogram obtained with reference solution (b) (1.0 per cent).
Liquid chromatography (2.2.29).
Test solution Dissolve 50 mg of the substance to be examined in the mobile phase and dilute to 100.0 mL with the mobile phase.
Reference solution (a) Dilute 1.0 mL of the test solution to 100.0 mL with the mobile phase. Dilute 1.0 mL of this solution to 10.0 mL with the mobile phase.
Reference solution (b) Dissolve the contents of a vial of valsartan for system suitability CRS (containing impurity C) in 1.0 mL of the mobile phase.
- — size: l = 0.125 m, Ø = 3.0 mm;
- — stationary phase: end-capped octadecylsilyl silica gel for chromatography R (5 µm).
Mobile phase glacial acetic acid R, acetonitrile R1, water R (1:500:500 V/V/V).
Flow rate 0.4 mL/min.
Detection Spectrophotometer at 225 nm.
Injection 10 µL.
Run time 6 times the retention time of valsartan.
Identification of impurities Use the chromatogram supplied with valsartan for system suitability CRS and the chromatogram obtained with reference solution (b) to identify the peak due to impurity C.
Relative retention With reference to valsartan (retention time = about 5 min): impurity C = about 0.8.
System suitability Reference solution (b):
- — resolution: minimum 3.0 between the peaks due to impurity C and valsartan.
- — impurity C: not more than twice the area of the principal peak in the chromatogram obtained with reference solution (a) (0.2 per cent);
- — unspecified impurities: for each impurity, not more than the area of the principal peak in the chromatogram obtained with reference solution (a) (0.10 per cent);
- — total: not more than 3 times the area of the principal peak in the chromatogram obtained with reference solution (a) (0.3 per cent);
- — disregard limit: 0.5 times the area of the principal peak in the chromatogram obtained with reference solution (a) (0.05 per cent).
Maximum 20 ppm.
Dissolve 1.0 g in a mixture of 15 volumes of water R and 85 volumes of acetone R and dilute to 20 mL with the same mixture of solvents. 12 mL of the solution complies with test B. Prepare the reference solution using 10 mL of lead standard solution (1 ppm Pb) R.
Maximum 2.0 per cent, determined on 0.500 g.
Maximum 0.1 per cent, determined on 1.0 g.
Dissolve 0.170 g in 70 mL of 2-propanol R. Titrate with 0.1 M tetrabutylammonium hydroxide in 2-propanol, determining the endpoint potentiometrically (2.2.20). Perform all operations under nitrogen.
1 mL of 0.1 M tetrabutylammonium hydroxide in 2-propanol is equivalent to 21.78 mg of C24H29N5O3.
In an airtight container.
Specified impurities A, C.
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10. Control of impurities in substances for pharmaceutical use): B.
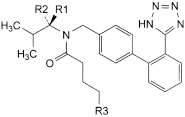
A. R1 = H, R2 = CO2H, R3 = CH3: (2R)-3-methyl-2-[pentanoyl[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]amino]butanoic acid,
C. R1 = CO2H, R2 = R3 = H: (2S)-2-[butanoyl[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]amino]-3-methylbutanoic acid,
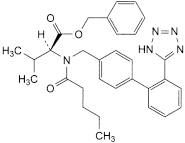
B. benzyl (2S)-3-methyl-2-[pentanoyl[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]amino]butanoate.
Ph Eur