



INTRODUCTION
Flow cytometry is an analytical method that plays a critical role in the quantitative and qualitative assessment of cell populations in patient and cellular product samples. The power of flow cytometry lies in its ability to rapidly and reliably analyze multiple attributes of individual cells within heterogeneous cell populations. Despite the value of flow cytometry data, method validation is challenging—perhaps more so than for other analytical methods—because of errors and artifacts from multiple sources.
Although flow cytometric methods can also be used to sort and isolate cells as part of the manufacturing process for cell- and tissue-based biological products, the scope of this chapter is limited to the use of flow cytometry as an analytical method. This chapter presents the technical aspects of the method, including instrumentation, sample handling and staining, and data analysis. Sources of error are considered in the context of technical features, as well as in the discussion of quality control, quality assurance, and standardization. Finally, current applications and assay troubleshooting principles are presented. For additional information on the basics and practical aspects of flow cytometry, see the current edition of Practical Flow Cytometry (Shapiro, 2003).
Flow cytometry is widely used to characterize cell and tissue-based products, but most assay methods are not yet standardized. In addition to issues related to technical complexity, there are also challenges to standardization of flow cytometric methods for specific product classes or types related to the heterogeneous nature of these products, even among those with similar manufacturing processes and clinical uses. Current and future innovations in instrumentation, analytic reagents, analytic algorithms, and automation are likely to improve the technology’s capabilities but are unlikely to eliminate challenges (e.g., bioassay, identification tests, and other applications).
PRINCIPLES OF OPERATION, METHODS, QUALITY, AND STANDARDIZATION
The process of flow cytometry requires that cells move past a fixed light source consisting of one or more lasers so that individual cells can be observed or interrogated for characteristics such as size, granularity, and presence of surface membrane or intracellular antigens or molecules. The cells are suspended in fluid in which movement is controlled by the size and configuration of tubing, chambers, and pumps specific to the flow cytometry instrument. The pattern of light signals produced from the laser light’s interaction with the cells is captured by a detection system, also specific to the instrument, and the detected signals are transformed into data elements that can be analyzed and combined with data from other cells in a given sample. Data from a cell suspension can then be expressed and presented in one-, two-, or three-dimensional visual formats, or in numerical formats, to characterize the cellular sample and its subpopulations both qualitatively and quantitatively.
Flow Cytometry Instrumentation
Flow cytometers, which incorporate fluidic, optical, and electronic signal processing elements, are described below.
fluidics
The fluidics system moves a bulk mixture of cells so that a stream of single cells is formed. Within the flow cytometer, the single-cell suspension passes through a confined region where each cell is sequentially illuminated by a uniform light source at the observation point (interrogation point). Most instruments use a flow chamber (flow cell) that, after the cell sample is drawn into the sample injection tip, combines the cells with isotonic sheath fluid, using a conical nozzle assembly that is geometrically designed to produce a laminar flow of fluid (Figure 1). The fluid outlet nozzle typically has an orifice of 50–250 µm in diameter through which fluid exits at a high flow rate. Differential pressures between the sample stream of cells (lower pressure) and the sheath stream (higher pressure) draw the cells/particles out into a confined stream. The resulting coaxial stream within a stream is highly efficient, and the sample stream at the observation point is typically only slightly larger than the cells or particles contained within. At least one manufacturer uses an alternative approach in which the coaxial stream strategy is replaced by the use of microcapillaries to focus and direct the cells. The light signal deflected or emitted by the cell is then measured and analyzed.
+'&img=../../images/v38332/c1027-f1.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 1. Schematic diagram of a flow cell.
optics
When cells are stained with fluorescent dyes or with fluorescent-labeled antibody reagents, light emitted from the laser interacts with the fluorescent dye to produce a stimulated emission that has coherent (parallel) waves of light of uniform wavelength, phase, and polarization. Fluorescent light signals generated from the interaction of the laser light with the cells are collected by an array of detectors oriented in direct line with, and at 90 to, the incoming laser beam. The most common commercially available flow cytometer lasers (with corresponding wavelengths) are the blue argon laser (488 nm), the red diode laser (635 nm), and the violet laser (405 nm).
to, the incoming laser beam. The most common commercially available flow cytometer lasers (with corresponding wavelengths) are the blue argon laser (488 nm), the red diode laser (635 nm), and the violet laser (405 nm).
electronic signal processing and data output
When a cell passes through the optical system of a flow cytometer, the light-scattering patterns or fluorescence from any fluorochrome on or in the cell are detected by various types of photodetectors or photomultiplier tubes (PMT) that transform the information about the characteristics of the cell into a computerized readout. Each analyzed cell generates an event in each parameter (forward scatter, side scatter, fluorescence) for which it is measured. Figure 2 shows an example of a typical two-color flow cytometer configuration. Different cell types have distinctive sets of signals in the various parameters. For example, when the cell passes through a beam of light, the light deflected in the forward direction (usually about 20 from the forward direction of the laser) is called forward scatter and is collected by a detector known as the forward scatter channel (FSC). The amount of deflection in the FSC is proportional to cell size. Light deflected at a 90 angle is known as side scatter and is collected by the side scatter channel (SSC). This provides a measure of the cell’s structural complexity caused by granules, membrane roughness, or nucleus, all of which are associated with higher SSC. The light deflected by other PMTs using a specific band-pass filter is collected by specific fluorescence channels (FL1 and FL2 in Figure 2). The electrical pulses, originating from light detected by the PMTs, are processed by a series of linear and log amplifiers. Logarithmic amplification is often used to measure fluorescence in cells. Figures 3–7 show histograms for cells stained with antibodies conjugated with specific fluorochromes (see Table 1). The antibodies are specific to some of the cluster of differentiation (CD) markers discussed in Immunophenotyping (see below).
+'&img=../../images/v38332/c1027-f2.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 2. Typical 2-color flow cytometer with detectors for FSC, SCC, and fluorescence.
Table 1. Fluorochromes Commonly Used
in Flow Cytometry
in Flow Cytometry
| Fluorochrome | Typical Excitation Laser (nm) |
Emission Peak (nm) |
|---|---|---|
| Cascade Blue | 375; 401 | 423 |
| Pacific Blue | 403 | 455 |
| R-Phycoerythrin (R-PE) |
480; 565 | 578 |
| PE-Cy5 conjugates | 480; 565; 650 | 670 |
| PE-Cy7 conjugates | 480; 565; 743 | 767 |
| Red 613 (Texas Red) |
480; 565 | 613 |
| Peridinin Chlorophyll (PerCP) |
490 | 675 |
| Fluorescein (FITC) |
495 | 519 |
| Allophycocyanin (APC) |
650 | 660 |
| APC-Cy7 conjugates | 650; 755 | 767 |
The versatility of flow cytometry comes from the ability to attach fluorescent tags to the cell’s surface, cytoplasm, or nucleus or to products of the cell. Fluorescent markers attached to the cell can be excited by lasers to emit light of specific wavelengths, and this light is then detected and analyzed in the manner described above. The type and amount of fluorescence detected provide both quantitative and qualitative information about the cell.
The photodetectors convert light into an analyzable output by generating a small current of which the voltage has amplitude proportional to the amount of light. The voltage is amplified and converted into electrical signals large enough to be plotted by the computer in several different ways. Thus, the FSC, SSC, and fluorescent detectors collect the light and convert it into electrical signals that can be analyzed by the computer. In this way, the signals coming from each photodetector can be measured for their intensity (low to high) and sorted into channels. The channels are arranged as a continuum so that a cell population with many large cells will have many events in the higher channels, and one with many small cells will have many events in the lower channels.
data analysis
Data output from the flow cytometer can be represented in several ways, the most basic of which is the single-parameter histogram (Figure 3), in which events with similar intensity of light (forward scatter, side scatter, or fluorescence) are collected in channels and then plotted. This plot demonstrates the number of cells with similar optical characteristics. Figure 4 is an example of graphs that display two measurement parameters, one on the x-axis and one on the y-axis, and the cell count as a density (dot) plot or contour map. The parameters could be SSC, FSC, or fluorescence. These parameters can be collected in one channel.
+'&img=../../images/v38332/c1027-f3.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 3. Single-parameter histogram showing expression of the cellular antigen CD3 in a mixture of cells.
+'&img=../../images/v38332/c1027-f4.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 4. Bivariate dot plot of cells displayed by FSC and SSC.
A dot plot displays a dot for each cell, and density plots and contour plots show a heat map or a topographical linear map, respectively, based on the relative number of cells in each channel. The forward versus side scatter histogram is the most common method of identifying different hematopoietic cell types. Figure 5 shows a contour plot that is a 3-dimensional representation of the relative number of cells in the various channels.
+'&img=../../images/v38332/c1027-f5.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 5. Bivariate contour plot showing relative numbers of cells present in each channel that co-express 2 CD markers.
When cells are stained with antibodies for different epitopes carrying two different fluorochromes, the data are presented as a plot of the two parameters plotted against each other. Cursors can be set on each axis to separate positive populations from negative populations for each of the attributes. This results in a graphic representation of cells that are positive for both markers, negative for both markers, and positive for only one of the two markers (Figure 6).
+'&img=../../images/v38332/c1027-f6.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 6. Schematic presentation of a 2-parameter histogram.
The flow cytometer allows the user to set the limits of positive and negative for each marker. Flow cytometric data are collected in list mode, where each electronic signal from a respective cell is displayed in the sequence in which it was acquired. List mode files can be edited at a later time to include or exclude any event. A basic advantage of flow cytometry is the ability to separate the data about the cells of interest from both the background and dead cells (e.g., noncellular particles or debris) when dealing with forward and side scatter and cells of other populations. The user must decide which signals are the actual light outputs from the cells and must construct an electronic gate to tell the computer to count as positive only the events that fall within the gate. Cell populations can vary widely depending on the tissue or cell source and the characteristics of the flow cytometer used. Gating allows the user to determine which outputs to consider actual events, so this process is of prime importance in standardizing flow cytometry data (Figure 7).
+'&img=../../images/v38332/c1027-f7-2s34.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 7. Gating of a cell population with low side scatter and high forward scatter, which is distinct from other cell populations in the sample.
A technique known as compensation can be used to separate spectral overlap of fluorochromes that have similar emission wavelengths. For analog-style flow cytometers manufactured before the late 1990s, compensation must be set before data acquisition. In modern digital instruments, compensation can be set either before or after data acquisition. The adjustment of compensation can be more of an art than a science, and considerable literature has been focused on the relative merits of various methods to determine the correct compensation for cell types or experimental conditions. The analyst should have considerable knowledge of the cell type under analysis in order to prevent errors in phenotyping that can result from improper adjustment of compensation.
The number of events counted should be adequate for statistical confidence in the results. The instrument can be set so that data are collected until a certain number of events in a channel have been measured. This feature allows the operator to vary the length of time or number of events from the sample so that statistically reliable data are generated. Thus, a sample that measures a rare event will analyze more total cells than one that measures a common event. Use of list mode files, the electronic data files that represent the most uncensored data, provides an advantage because these files can be further analyzed after data acquisition. Investigators need to ensure that all raw data, documentation, protocols, specimens, and final reports are archived at the close of the study. To ensure the integrity and quality of raw data collected, researchers need to abide by U.S. FDA Regulations for Good Laboratory Practices (GLP) as prescribed in 21 CFR Part 58, Good Laboratory Practice for Nonclinical Laboratory Studies; and Part 11, Electronic Records and Electronic Signatures.
Flow Cytometry: Elements of a Procedure
Flow cytometric methods incorporate sample handling, preparation, and staining; instrument setup and operation; data collection, analysis, and storage; and associated quality control measures.
sample handling and staining
Sample Collection, Handling, and Anticoagulation
In order to make accurate conclusions about the cell-based drug product, the analyst should ensure that samples from cellular products are as representative as possible of the whole product. Blood, apheresis samples, and cell suspension products should be well mixed before sampling, and care should be taken to obtain adequate sample volumes.
Cellular samples containing human blood/plasma must be anticoagulated. Citrate-based anticoagulants (e.g., Anticoagulant Citrate Dextrose Solution A) or heparin are recommended more highly than EDTA because they will optimally preserve samples being held for more than a few hours. For longer-term samples, specific transport/storage media may be required, and validation studies should be performed to ensure that those samples are equivalent to fresh samples at the time of flow cytometric analysis.
Samples intended for whole blood lysis and surface antigen staining should be transported and stored, preferably on a slow oscillating mixer, at room temperature. Fixed samples or live cell preparations should be stored at 4. When the sample may be exposed to extreme temperatures, temperature-control materials (room temperature packs, wet ice/cold packs, and insulation) may be necessary, and validation studies should be performed to ensure sample integrity. For critical or high-value samples, temperature-monitoring devices may be needed during transport.
After acquisition, specimens should be analyzed as soon as possible. Special attention should be given to situations in which cellular proliferation and metabolic depletion of energy sources within the transport/storage media can lead to apoptosis. When accurate counting is not required or if infectious agents are suspected, a commercial lyse/fix solution can increase storage time and reduce the risk of disease transmission. For specimens separated by density-gradient centrifugation, storage in a solution of buffered paraformaldehyde (0.1% to 2.0%) is recommended after cell labeling has occurred.
Sample Processing, Staining, and Fixation
Reagents used in sample processing, staining, and fixation should be qualified for their intended use. Further guidance is available in ICH Q6B, Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products. When using reagent kits, follow the manufacturer’s sample processing instructions.
Sample processing that involves centrifugation, washing, red blood cell (RBC) removal or lysis, or density-gradient separation is commonly done during many flow cytometric applications but can introduce error and artifact. Several techniques and reagents are available for RBC removal and lysis. Clinical grade [in vitro diagnostics (IVD) or analyte-specific reagents (ASR)] reagents are recommended for optimal quality, but artifacts can still occur. Density-gradient centrifugation can introduce error associated with variable cell losses among subpopulations that are being measured. These sources of error and artifact can be avoided by analyzing live whole blood whenever possible.
Most whole blood lysate instructions recommend staining at room temperature and in the dark. Many methods include a dilute fixative to prevent capping and internalization of fluorochrome. In contrast, cell preparations (density-gradient cell preparations, apheresis specimens, tissue culture) should be stained at 4, washed with cold buffer, and stored cold until analyzed.
Fixation that also preserves cell surface antigens can be accomplished using commercial leukocyte preservatives or with buffered formaldehyde or paraformaldehyde. Very little validation of storage times, antibody binding, or fluorochrome intensity has been reported. Any laboratory that considers batch analysis of fixed specimens should validate these techniques thoroughly before implementing.
use and choice of fluorochromes
Fluorochromes
Fluorochromes are used for direct staining of cells or as agents bound to antibody or other reagents to stain cellular antigens or other structures. Table 1 lists examples of common fluorochromes used for flow cytometry and their excitation and emission wavelengths. Wavelengths (nm) may vary slightly depending on the environment. Synthetic probes from specific manufacturers are also available.1 Fluorochromes must match the spectral range for the lasers and filter sets specific to the user’s flow cytometer.
When choosing fluorochromes for multicolor phenotyping, the operator should refer to established methods for the particular instrument. In general, the brightest fluorochromes should be matched with the antigens that are expected to have the lowest expression on the cell surface. The brightness of tandem dyes can also be reduced by the use of certain fixatives, some of which are less problematic than others. When designing a multicolor flow and tandem dye procedure that has not been previously validated, the operator should consult the manufacturer’s technical service, compare tandem dye/fixative combinations, and validate the final fluorochrome combination to ensure sample-to-sample consistency.
Tandem fluorescent dyes are dual-conjugated fluorescent molecules. When the two labels are in close proximity, energy produced by the laser exciting the donor fluor is transferred to the acceptor fluor, releasing a photon at the emission wavelength of the acceptor fluor (also known as fluorescence resonance energy transfer, or FRET). For example, PECy5 will excite at the excitation wavelength for PE (565 nm), transfer energy to Cy5, and emit at the emission wavelength for Cy5 (670 nm).
Fluorescently Labeled Antibodies
Most commercially available antibody reagents are monoclonal, but polyclonal reagents may be available, and desirable, for some applications. The quality and specificity of an antibody can vary widely. Antibodies directed at a given antigen may differ in their binding specificity for different antigenic epitopes or in the strength of binding to the same epitope. If possible, use directly conjugated fluorochrome–antibody combinations that are IVD or ASR grade. Optimization of antibody concentration for the desired cell population is protocol specific but is generally accomplished by using increasing concentrations of antibody with a fixed number of cells to bracket the optimal brightness between autofluorescence and quenching. Quenching is caused by the prozone phenomenon, which occurs when excess antibody leads to immunoprecipitation and loss of fluorescence intensity. Further details are available in methods manuals such as Current Protocols in Immunology (Coligan et al. 1994).
Cell Surface Antigen Staining
Techniques for surface antigen staining vary with the type of specimen. Whole blood lysis techniques generally require surface labeling at room temperature in darkness for 15–30 min, followed by RBC lysis and, if desired, fixation. Published techniques use ammonium chloride (NH4Cl) lysis of whole blood or marrow specimens followed by washing before antibody labeling for leukemia immunophenotyping. Mononuclear cell or cultured cell samples that are stained live should be kept at 4 or in an azide-containing buffer to prevent capping and internalization of the antibodies.
Intracellular Staining
Several standardized procedures also exist for labeling intracellular antigens and cytokines. The operator should consult the manufacturer’s protocol and standardized reagents for these procedures. Because permeabilizing reagents vary among procedures and manufacturers, do not mix and match reagents. For cytokine labeling it is often necessary to use an activating step and a Golgi block to allow a sufficient amount of cytokine to accumulate for detection. If standardized reagents or procedures are not available from the manufacturer or if analysis of specialized functions is required, many common procedures and techniques can be found in sources such as Current Protocols in Immunology (Coligan et al., 1994).
Quantitation of Antigens
Some applications require quantitation of the average number and density of antigen molecules per cell in order to give a more complete picture of the immunological behavior of cells (e.g., in studies where extracellular antigens are expressed differentially in relation to activating stimuli). The intensity of an antibody/fluorochrome labeled cell preparation is compared to the intensity of a set of microbead fluorescence standards collected at the same PMT voltage settings. The standards are calibrated in molecules of soluble fluorescence (MESF) units, from which one can determine effective fluorescence to antibody (F/P) ratio, the number of antigen molecules per cell, and the density of available sites per cell.
instrument setup and operation
Compensation
Most instrument manufacturers supply software and test reagents (usually fluorescent beads) to set PMTs and compensation in order to target values found with the most common clinical tests (e.g., lymphocyte phenotyping, CD34 analysis). The operator should also use a biological control such as preserved blood or mononuclear cells, which are commercially available. Compensation must be set before acquisition on analog instruments. Digital instruments’ PMTs must be set correctly because the values for these settings cannot be changed once the list mode file has been generated. When rare events are examined and/or intracellular dyes (e.g., 7-amino actinomycin D [7-AAD], propidium iodide [PI], Syto-16, etc.) are used in conjunction with fluorescently labeled antibodies, the balance of PMT voltage and spectral overlap must be closely monitored.
Autofluorescence (AF) is fluorescence above baseline in the absence of fluorochrome staining. This occurs in some cells, typically myeloid cells (especially alveolar macrophages) and cultured primary cells. If desired or necessary, AF can be measured directly on a fixed PMT voltage or can be calculated from a reference standard of fluorescent reference bead preparations (see Quantitation of Antigens, above). Avoid use of the 488 or 532 nm excitation wavelength and subsequent spectral compensation of the AF as an additional fluorochrome.
Data Acquisition and Gating Strategies
When possible, all events should be acquired in list mode, i.e., without selective gating of events. Live gating, defined as selective gating of events during acquisition, should be employed only when the desired subset is sufficiently rare that >2 million total events must be analyzed in order to count a significant (100 or greater) number of events of the desired population. List mode data can be acquired uncompensated when digital instrumentation is used, but most operators find that analysis is much less difficult and time-consuming if the data are in the range of proper compensation before acquisition. In addition, it is often desirable to set thresholds for exclusion of debris. Setting a forward scatter threshold, for instance, excludes events below a predetermined size in order to prevent the large list mode file size that can occur when these events are counted.
Use of Controls
Fluorochrome-conjugated bead preparations are used for standardizing PMTs and compensation and for quantifying the expression of specific markers. The use of biologic controls is also highly recommended. Stain the cell samples with an isotype control and primary and secondary antibodies to assess nonspecific binding unless the laboratory has ascertained by rigorous validation procedures that nonspecific binding does not interfere with assay results.
Antigen-positive and -negative cell populations (prepared and stained in a manner identical to that for the test articles) provide internal system suitability standards. Such control cell populations also allow the laboratory to assess lot-to-lot variations in antibody preparations and staining reagents.
Use of Dyes and Gating for Cell Viability
Cell viability dyes such as 7-AAD, PI, and TO-PRO iodide are commonly used to determine the proportion of dead cells in a cell therapy product. These dyes are typically excluded from live cells but pass through the cell membranes of dead cells, staining their DNA. Cell viability staining can be combined with surface membrane or intracellular staining to evaluate subpopulations and the proportion of live and dead cells stained with a given marker. Viability staining can also be used in conjunction with a membrane dye in flow cytometry-based cytotoxicity assays. These viability dyes should not be confused with the many apoptosis-detection reagents now available. Validation techniques for non-IVD viability dyes involve the preparation of a dead cell population that is added in serial dilution to a live cell product, and the cell mixture is then assessed for fidelity to the known proportion by staining with the dye of interest.
Cell Enumeration
Absolute cell count, expressed as the number of cells in a given sample volume, can be determined by dual- and single-platform methods. The dual-platform method relies on a separate automated cell-counting instrument or manual counting method to first enumerate the cell population. The percentage of a subset(s) of interest is then determined by flow cytometry, that percentage is multiplied by the cell count, and the result is divided by 100. Single-platform methods enumerate the cell population and subset counts directly by counting the cells in the sample simultaneously while counting reference beads that have been added to that sample volume in a known concentration. Reference beads are often provided as a bead suspension that is added to the specimen. Alternatively, a given volume of sample may be added to a known number of reference beads provided as a solid phase matrix in polystyrene tubes. These approaches are subject to pipetting error, so extra care must be taken to ensure accuracy and reproducibility.
Instrument Setup and Quality Assurance
Each laboratory should have a quality plan that defines the standard operating procedures for instrument setup and calibration, as well as regular instrument monitoring, maintenance, and cleaning. Instrument logs should document these activities and operators. In general, the instrument manufacturer’s quality program should be followed unless a suitable alternative has been established.
Instrument parameters such as laser current, voltage, output, and PMT voltages during calibration should be monitored and recorded whenever the instrument is in use. Careful monitoring of instrument setup parameters can be helpful in detecting trends and predicting laser or PMT failure. Biological control testing results should also be monitored and recorded to detect and prevent analytical method drift.
The laboratory should also participate in a proficiency-testing program that reflects the test menu. Depending on need, this could range from a formal program such as the one administered by the College of American Pathologists (CAP) to simple sharing of specimens and analysis with another laboratory. Ensuring that operators are trained, qualified, and periodically evaluated for proficiency to perform specific procedures will also help ensure the consistency of techniques and controls.
data management and statistical considerations
Data Management and Storage
Quality control assays and sample test assays (in list mode) should be stored in a manner that complies with regulatory requirements applicable to the laboratory. This can be accomplished by transfer to fixed drives, removable media, or to a server such as a commercial laboratory information system. Storage of results should always be traceable to the original FCS list mode file, instrument settings, and quality control parameters for that particular specimen. Data should be backed up to avoid loss of files. Storage and backup procedures should also be established for manual (paper) records that may be used for calculations and summary data.
Data Analysis and Statistical Considerations
For most flow cytometry applications, data analysis involves displaying the data from list mode files or live gating in a plot (single-parameter histogram plot, two-parameter dot plot with regions, or three-dimensional plot), and measuring the distribution of events within that plot. Further analysis of data within selected populations can be done by gating on specific cell populations. Description of the data typically includes the percentage of events within the population with a given characteristic (forward scatter, side scatter, fluorescent marker). The numerator is the number of events with the characteristic, and the denominator is either the number of total events counted or the number of gated events counted. For two-dimensional plots, analysis is typically done using computer software that analyzes and reports regional (e.g., quadrant) statistics. Because cell population clusters may shift their positions from one data file to the next, software has been developed for cluster analysis.
Statistical analysis of quantitative flow cytometry applications differs from qualitative applications in which a cell is considered either positive or negative for a given marker. For a typical quantitative application in which the number of molecules on the cell surface is estimated, the mean or median fluorescence intensity of the sample cells labeled with a fluorescent antibody bound to the molecule of interest can be compared to appropriate controls, including standard curves of cells or particles with known quantities of that molecule/antibody.
A common practical consideration for flow cytometric analysis of cell therapy products, especially autologous and related donor allogeneic products, is that sample size for analytical testing is often limited because of the limited cell content of the therapeutic product itself. This creates special challenges if cells containing the flow marker of interest are rare events. In these cases, before making a decision about sample size, the user must consider the expectations for detecting the rare events within a given number of total cells (i.e., prevalence, variability, sampling error) in relationship to the desired precision of the estimate.
Quality and Standardization
Standardization of flow cytometry practices and equipment requires validation, quality assurance (QA), and quality control (QC) practices. Although flow cytometry is used widely in both research and clinical laboratories, testing of cells for the development of clinical diagnostic and therapeutic applications is increasing, leading to more comprehensive regulatory requirements and attention to standardization. As an example, flow cytometry operators have traditionally used fluorescent microspheres (beads) or cells for instrument setup and QC, frequently based upon manufacturers’ recommendations. However, consistent instructions about how these control standards should be applied to instrument setup and QC remain elusive.
Properly applied, validation provides documented evidence that the manufacturing or testing process consistently produces product that meets predetermined specifications. Based on a thorough understanding of critical process parameters, validation helps to define product quality and helps to ensure a consistent and well-controlled manufacturing or testing process. Validation of flow cytometric methods should incorporate instrument qualification, analytical method validation, and operator qualification.
documentation
GLP and GMP processes require appropriate documentation such as standard operating procedures (SOPs) for all lab processes. SOPs must also be periodically updated and approved to reflect current practices. Training and qualification are required so that laboratory staff have the appropriate level of competence for their assigned responsibility. Operator competencies must be continually reviewed and assessed in relationship to SOPs and policies.
Integrating both internal and external quality processes is an important element of quality assurance. These involve equipment validation, manufacturing controls and limits, and product specifications. Process and equipment validation processes generally require installation qualification (IQ), operational qualification (OQ), and performance qualification (PQ).
equipment and assay qualification
IQ establishes that the instrument is received as designed and specified, and that it is properly installed in a suitable environment. This generally means checking physical and facility requirements to determine whether the flow cytometer can be suitably installed. Qualification factors verified typically include temperature, humidity, space, and electrical facility capabilities in relationship to the instrument manufacturer’s requirements. IQ procedures also ensure that all hardware and software components purchased are installed properly by the instrument manufacturer’s representative.
OQ demonstrates that an instrument will function according to the manufacturer’s specifications. This generally means component-level testing by the instrument manufacturer’s representative or using a manufacturer’s validation package that guides the end user to perform this function. Where possible, these tests should have specifications with corresponding quantitative control limits. This testing ensures that the instrument hardware and software are operational by comparison with the manufacturer’s specifications.
PQ demonstrates that both the instrument and the assay consistently perform according to specifications. For flow cytometry, these specifications are generally determined by the laboratory performing the flow cytometric testing and usually include daily instrument and assay control test specifications. For specific assays, PQ should incorporate standardized methodology, application-specific setup and compensation, and specifications for linearity, precision, and accuracy of reported assay results. On some digital flow cytometers, a baseline instrument setup may be necessary in order to determine the optimal instrument settings for a given assay.
instrument performance
Performance specifications may be identified by the flow cytometer manufacturer and may not address all of the operator’s specification requirements. After identifying the manufacturer’s specifications, the laboratory must establish specifications that are appropriate for the hardware configuration(s) that will be used, and the specifications must be standardized. As an example, the manufacturer may have a base specification for a four-color dual-laser flow cytometer. If the base specification calls for the standard red diode 635-nm laser but a helium–neon air-cooled 633-nm laser is substituted, the base specifications are no longer valid for the system. Similarly, if the specifications are based on the use of a 660-nm band-pass filter but a 675-nm long-pass filter is substituted, the base specifications are not valid because of differences in the emission filter characteristics.
performance monitoring
Instrument performance can be monitored on a daily basis, using commercially available fluorescent beads. Light detectors such as PMTs, photodiodes (PD), and avalanche photodiodes (APDs) are used in most systems to detect signals, and their gains can be changed to increase or decrease the sensitivity of the detectors. Therefore, monitoring the settings of these detectors is as important as monitoring the signals, and these settings should be associated with each raw data file. The easiest approach is to maintain the same instrument detector settings from day to day and to measure the intensity of the fluorescence signals. This approach should be implemented for all parameters that must be validated using the appropriate beads. Instrument sensitivity is based on the ability of the detection system to resolve dim cell or bead populations. For this reason, measuring the coefficient of variation (CV) of dim to moderately intense fluorescent bead populations is a means to monitor fluorescence sensitivity on a daily basis. The instrument manufacturer’s recommendations should be used to monitor performance.
Ambient high temperature can affect laser and PMT performance and should also be monitored on a daily basis.
Incorrect compensation for spectral overlap can strongly affect data during multicolor analysis. Many approaches have been established for this purpose, and recently mathematical algorithms have been used rather than analog circuitry. Algorithms, such as those that use matrix algebra, enable the operator or investigators to apply objective criteria to compensate for spectral overlap after all data have been collected. On older systems, the standard approach has been to compensate using a subtractive hardware adjustment to the observed preliminary data before all data have been collected. This approach can be subjective and is not as likely to produce accurate results as compensation by newer methods. Antibody-bound capture beads are valuable compensation tools because they can be used with the same antibody and tandem dyes for all fluorophores that will be used on cells. Validate the use of beads in place of cells for compensation purposes.
standards
Microsphere-based fluorescence standards for flow cytometry have been categorized by their purpose:
-
Type I standards are alignment standards that are used to make adjustments to the instrument’s optical alignment. These are typically used by field service engineers and by users of operator-adjusted systems to check the optical signal alignment in order to improve instrument sensitivity. These particles are typically small (
 2 µm) and bright, and they provide the most uniform illumination.
2 µm) and bright, and they provide the most uniform illumination.
- Type II standards are reference beads and are the most commonly used bead standards. These typically are used on a daily basis, have dim to moderate fluorescence intensity, and can be obtained with various attached fluorophores. These can be used to mimic cells and, with dedicated software, to determine relative instrument sensitivity.
- Type III standards are used for fluorescence calibration. These are used for specialized applications that require calibration of one or more fluorescence detectors for quantitation of molecules of fluorochrome. Determination of the ratio of fluorophores to antibody (F/P ratio) allows subsequent calculation of the number of antibodies bound per cell.
instrument setup and quality assurance
Instrument setup and quality assurance should be activities independent of the biological assay. Many variables related to instrumentation can lead to artifacts in the biological assay results. Two activities can be used for instrument quality assurance: baseline setup and daily setup.
Baseline Setup
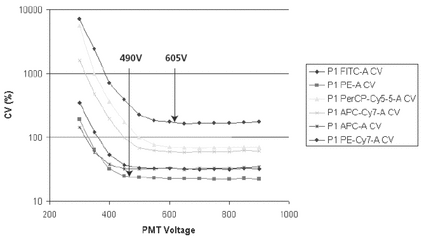
On newer digital instruments it is desirable to establish a baseline setup of instrument settings that provide optimal sensitivity. This setup is not a daily procedure but should be performed if the instrument configuration is changed or if the instrument is serviced. Because PMT voltages and instrument configuration can strongly affect instrument sensitivity, this method should be used to provide objectivity as well as improved sensitivity. PMT voltages can be increased to a range that provides a lower CV (Figure 8). These settings can be, but may not always be, used for the biological assay.
+'&img=../../images/v38332/c1027-f8.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 8. PMT voltages can be increased to a range that provides a lower CV.
Daily Setup
Type II standards, as reference particles, can be used to monitor signal intensity, separation of moderately bright and dim particles, and signal resolution. Fluorophore-matched beads provide a compensation tool as well as a means of knowing that the instrument is able to detect those wavelengths. Fluorophore-matched beads, however, do not have the fluorescence uniformity required for measuring CVs. Based on the optical performance of the instrument, CVs are best measured using dim and moderately bright hard-dyed beads such as coumarin-dyed microparticles that fluoresce in a broad spectral range.
It is easy to confuse assay controls and instrument controls. Beads provide a fluorescence uniformity and consistency that cannot be obtained with cells, and, accordingly, beads are useful for monitoring instrument performance. For this reason, it is better to use a process control cell preparation to verify compensation and fluorophore acceptability.
Instrument settings for daily setup are generally the same settings used for assays. Not all assays can be used with the same instrument settings, and it is not always necessary to perform these activities for every instrument setting unless it is required by validation.
Daily activities include consistent instrument setting from day to day, use of a broad range of dim to moderate intensity beads, and monitoring key parameters, including bead fluorescence intensity (absolute and % CV), PMT values, temperature, laser power, and laser wavelength.
assay quality control and quality assurance
Assay-specific instrument settings should be established to demonstrate that all cell populations can be identified in bivariate plots for fluorescence and light scatter. Most importantly, positive populations must be on scale and properly compensated. This is critical when exciting cells with red lasers, which do not cause cells to generate significant autofluorescence. For this reason, it is important to verify appropriate PMT settings so that the positive population is in the upper part of its fluorescence scale, because it may be extremely difficult to identify the negative cells.
Fluorescence compensation is a critical adjustment. Digital instruments provide objective offline adjustments during analysis, and detailed instructions for proper compensation settings are available. Using cells or capture beads stained with a single antibody-fluorochrome is generally the best approach, but specialized fluorochrome-labeled bead mixtures can also work well to compensate for multicolor acquisition and analysis.
isotype controls
An isotype control is a negative-control antibody that should not react with the antigen of interest and is the same isotype as the test antibody. Myeloma protein or immunoglobulin that has no specificity to the species being tested and has the same Ig chain class and subclass as the test antibody is conjugated to a fluorochrome identical to that on the test antibody. Ideally, very little or no binding occurs when the isotype control is used in parallel with the test. Idiotypic nonspecific binding frequently occurs, however, and is independent of the isotype of the antibody. This is most likely related to other differences in antibody chemistry and can be especially problematic with rare-event detection assays, such as those for hematopoietic stem cell assays in peripheral blood.
fluorescence minus one controls
Fluorescence minus one (FMO) controls are used to control nonspecific staining during a multicolor assay. After compensation has been set, a tube containing all of the fluorochrome-labeled antibodies, except one, is run. If the compensation has been properly set, any positive fluorescence in the parameter corresponding to the missing fluorochrome-labeled antibody is caused by nonspecific staining and can be an indication of antibody excess or degradation of related tandem dyes. Although FMO controls are very useful for estimating the sensitivity of a particular detector in the context of other reagents, the controls do not take into account nonspecific binding that can occur with the addition of the test antibody. FMO control tubes are most appropriately used for troubleshooting or when establishing a new multicolor reagent cocktail.
process controls
Process controls, also known as system suitability standards, account for sample preparation and data acquisition. They can include commercially available preserved control cells, cell lines, or primary cells such as normal peripheral blood. Process controls can also be used to test new lots of antibody reagent against old lots.
biological controls
When treated or stimulated cells are compared to untreated or unstimulated cells, the untreated or unstimulated cells may in some cases be the most useful control for setting a positive/negative boundary. However, use of isotype controls may also apply to these situations, because stimulation may lead to Fc receptor upregulation, leading in turn to increased background staining, the presence of which can be elucidated by an isotype control.
FLOW CYTOMETRY APPLICATIONS FOR CELLULAR SAMPLES AND CELL THERAPY PRODUCTS
A wide variety of flow cytometry applications have been developed for research, clinical diagnosis and monitoring, drug development, and cellular product characterization and quality assessment (i.e., control to allow batch release). Traditional clinical applications include monitoring HIV disease and diagnosis and monitoring leukemia and lymphoma. Both pharmaceutical and academic research laboratories have increasingly broadened the application of flow cytometry from immunophenotyping to functional cellular assays, as well as microsphere-based multiplex assays capable of measuring multiple functional parameters on individual cells. Current functional assays include those that allow direct study of cellular activation status by measuring intracellular cytokine production or secretion of chemokines or cytokines, using a ligand-binding sandwich assay on microspheres.
Immunophenotyping
Flow cytometry allows the characterization of leukocyte subtypes by labeling cells with fluorochrome-conjugated monoclonal antibodies. The CD system defines monoclonal antibodies that recognize unique cell-surface antigens. Many clinical applications take advantage of flow cytometry’s unique capabilities to measure multiple CD antigens on thousands of individual cells.
cd4 enumeration
In the early 1980s, investigators discovered that HIV infects CD4 T cells and that a patient’s peripheral blood CD4 T cell count is a useful indicator of immune status. CD4 enumeration has become the most commonly used diagnostic test in HIV-infected patients to determine the need for anti-retroviral (ARV) therapy and for monitoring the effectiveness of ARV drugs. T cell subset counts are typically expressed in terms of cells per microliter and as a percentage of lymphocytes, using a standardized reagent, software, and instrument system.
leukemia and lymphoma
Multidimensional flow cytometric analysis enables identification of aberrant cell populations in bone marrow, lymphatic tissue, and peripheral blood of patients with leukemia or lymphoma. This is accomplished with oncology-relevant and lineage-specific cocktails of monoclonal antibodies. With optimal fluorophores and improved optical/electronic configurations in flow cytometry instrumentation, additional cell markers can be detected to more precisely identify leukemia or lymphoma cell phenotypes and to improve the physician’s assessment of patient status. Rare-event detection methods have improved the ability to detect minimal residual disease.
dendritic cells
Dendritic cells (DCs) act as antigen-presenting cells that can influence the nature and strength of the immune response to specific antigens. This finding has led to the development of DCs as cell-based therapies for cancer, infectious disease, and autoimmune disease. DCs are morphologically and phenotypically diverse and can be derived from several cell types. Two major DC lineages, known as myeloid and plasmacytoid DCs, can be segregated on the basis of their expression of CD11c and CD123, respectively. Additionally, the expression of the costimulatory molecules CD80 and CD86 can be monitored to determine DC maturation state.
stem and progenitor cells
CD34 expression is commonly used to characterize hematopoietic stem cells (HSCs) in peripheral blood, cord blood, bone marrow, and purified HSC preparations from these sources. Flow cytometric identification and enumeration of HSCs is possible by using monoclonal antibodies specific to the CD34 class III epitope, along with other well-characterized reagents, analysis software, and protocols. The reagent combination of anti-CD45, anti-CD34, and a viability dye such as 7-AAD is widely used for clinical applications. Increasing interest in developing cell-based therapies from embryonic, fetal, and adult tissue sources has led to the use of a wide variety of conventional and novel phenotypic markers for characterization of source cells and their more differentiated progeny. Flow cytometric assays are being developed as part of assay batteries to assess differentiated cellular products derived from pluripotent stem cell sources. These assays will help define appropriate numbers and types of desired cell populations, as well as help detect undesired cells such as residual pluripotent cells that could prove tumorigenic in the recipient.
leukocytes
Leukoreduction of blood products is a process used to produce blood products with a residual leukocyte content of less than 5 × 106 per unit. Clinical data suggest that nonhemolytic febrile transfusion reactions can be prevented by leukodepletion. Leukodepletion also prevents alloimmunization to HLA antigens in patients who will repeatedly require transfusion of blood products. Flow cytometry is routinely used to quantitate leukocyte contamination in leukocyte-depleted blood products.
platelets
Flow cytometry is a rapid and useful method for diagnosing many primary thrombocytopathies related to defects in structural or functional glycoproteins (e.g., abnormal expression of gpIIb/IIIa in Glanzmann thrombasthenia or gpIb in Bernard-Soulier disease). The use of thiazole orange, a fluorescent dye that binds RNA, allows immature platelets (reticulated platelets) to be quantified. The reticulated platelet count can be used to determine the rate of thrombopoiesis. This measurement can separate unexplained thrombocytopenias into those with increased destruction and those with defects in platelet production.
erythrocytes
Rhesus D-negative women receive prophylactic Rh-immunoglobulin to prevent alloimmunization from Rh(D)+ erythrocytes (RBCs). If fetomaternal hemorrhage is suspected, the mother’s blood is tested for the presence and quantity of fetal RBCs, using fluorescently labeled antibodies to the Rh(D) antigen or to hemoglobin F.
The reticulocyte count is used to help determine whether the bone marrow is responding adequately to the body’s need for RBCs and to help classify different types of anemia. Reticulocyte counts are based on the identification of residual ribosomes and RNA in immature nonnucleated RBCs. Flow cytometric enumeration of reticulocytes and their discrimination from mature RBCs uses fluorescent dyes that bind the residual RNA (e.g., thiazole orange).
Bead-Based Immunoassays
Multiplex microsphere-based flow cytometric assays combine a series of particles of discrete size and/or fluorescence intensity with matched antibody pairs to allow simultaneous detection of multiple soluble analytes on a flow cytometer. The flow cytometer’s capacity to discriminate particles on the basis of size and color enables determination of multiple results from a single tube or well. Many investigators use such assays to measure secreted chemokines or cytokines, kinases, and anti-HLA antibodies.
Proliferation Assays
dye incorporation into dna
Bromodeoxyuridine (BrdU) is a thymidine analog that can be incorporated into the DNA of cells during S phase, and then can be detected using specific, labeled monoclonal antibodies. By pulsing a stimulated cell culture with BrdU, cells can be identified that have proliferated (passed through S phase) during the time of the pulse. This assay has become a useful alternative to 3H-thymidine incorporation as a measure of proliferation because it is nonradioactive and can identify phenotypes of proliferating cells by the use of multiple markers and flow cytometry.
dye incorporation into cellular proteins or cell membrane
Cell-tracking dyes such as carboxyfluorescein succinimidyl ester (CFSE) and PKH26 have proven useful in the assessment of cell proliferation. CFSE binds covalently to cytosol and membrane proteins, and PKH26 binds non-covalently to cell membranes. When cells divide, CFSE/PKH26 labeling is partitioned equally between the daughter cells, which are therefore half as fluorescent as the parents. The fluorescence of each cell is further halved with each succeeding generation. This property makes CFSE/PKH26 assays useful not only for determining the fraction of cells that have proliferated in a stimulated culture but also, under ideal conditions, the number of generations that have elapsed. In this manner, the precursor frequencies of small populations that have proliferated over several days in culture can be calculated.
Functional Assays
intracellular cytokine expression
Cell surface and intracellular labeling techniques have been applied to the identification of cell subsets with specific functional characteristics. For example, brief stimulation of cells such as PBMC with protein or peptide antigens can result in the expression of activation markers and cytokines that can then be measured along with other phenotypic markers on the surface of the responding cells. The use of a secretion inhibitor such as brefeldin A or monensin allows the intracellular accumulation of cytokines. The cells are then fixed, permeabilized, and detected by a flow cytometric method. Such assays are useful for monitoring T cell subpopulations that respond to vaccines, infectious disease agents, or cancer. Functional properties of other cell types, including monocytes, DCs, and NK cells, can also be monitored using functional assays with appropriate stimuli.
kinases
Phosphorylation-specific cell activation intermediates can be identified using phospho-specific antibodies and flow cytometry. These reagents are useful for mapping intracellular signaling mechanisms, often in the context of other cell-surface phenotypic markers. Thus, multicolor flow cytometry can provide single-cell assessment of intracellular activation states in complex cell populations. These assays may have utility in detecting altered signaling states in cancer cells or in directing appropriate therapies based on the signaling properties of a patient’s tumor cells.
apoptosis
Apoptosis, commonly described as programmed cell death, is the process of cell death caused by regulated, physiologic processes. The apoptotic process manifests itself as a series of morphological, biochemical, and molecular changes to the cell and can be initiated by external or internal stimuli. A central event during apoptosis is the activation of caspases, a family of proteolytic enzymes. Caspases are synthesized as inactive proenzymes and are activated by other caspases or by similar molecules. They form a cascade that can lead to the cleavage of various cytoplasmic or nuclear proteins. One of the caspases that is reported to be crucial for the apoptotic process is caspase-3, which is activated during the early stages of apoptosis.
Flow cytometric methods for detecting apoptotic cells include measuring morphology, changes in membrane structure, DNA cleavage by endonucleases, and mitochondrial membrane potential. Natural or artificial caspase substrates or antibodies against the activated form of the enzyme have also been used for this purpose.
cell viability
Flow cytometry is often used to discriminate live cells from dead cells. The principle of nucleic acid dye exclusion is the basis of this application. A nucleic acid dye such as PI or 7-AAD is added to cells in suspension. During flow cytometric analysis, cells that fluoresce above background are considered nonviable because they cannot exclude the dye, which fluoresces when it binds to cellular DNA.
Flow Cytometry Immunoglobulin Assays
flow cytometry cross-matching
Before organ transplantation, flow cytometry cross-matching (FCXM) is performed on recipients to screen for anti-HLA antibodies that can cause rejection. Anti-HLA antibodies are detected by incubating HLA-defined leukocytes, B-cell lines, or HLA antigen-coated beads with the serum sample, followed by anti-human immunoglobulin fluorescently labeled antibodies. Leukocytes are immunostained to identify T and B cells in order to distinguish between anti-HLA class I and II activity, respectively. In addition, screening of blood donations for anti-HLA antibodies is also increasingly employed to identify donors whose blood products may have increased risk of causing transfusion-related acute lung injury (TRALI) in recipients.
anti-human neutrophil antibodies
Anti-human neutrophil antibodies (HNA) can cause neutropenia and have been implicated in TRALI. Autoimmune neutropenias may develop in patients who have autoimmune disorders such as Felty syndrome, systemic lupus erythematosus, and Hashimoto thyroiditis. The absence of anti-HNA antibodies narrows the differential diagnosis to nonimmune causes such as bone marrow failure, myelodysplasia, or marrow-infiltrative processes. Flow cytometry can detect anti-neutrophil antibodies and can confirm the origin of neutropenia or TRALI.
anti-human platelet antibodies
Anti-human platelet antibodies (HPA) are detected by both indirect and direct flow cytometry-based platelet-associated immunoglobulin assays. In autoimmune thrombocytopenic purpura, free serum antibodies are not found as frequently as are platelet-bound antibodies. In cases of alloantibody formation, serum antibodies may be detected without evidence of platelet-associated antibodies.
FLOW CYTOMETRY ASSAY TROUBLESHOOTING
When developing a flow cytometry method, first determine the ultimate purpose of the assay. For assays intended for research, the cell samples, reagents, and protocols may be difficult to standardize. Assays intended for patient diagnosis or to qualify a cellular product for release before administration to a patient demand more stringent assay and sample standardization. Regulatory guidelines, the type and stage of clinical investigation, and the ultimate purpose of the assay determine the level of assay rigor required.
Flow cytometry assay development should include the establishment and qualification of staining, handling, instrument, and analysis parameters and limits. Assuming that the method has been well developed, the operators are properly trained, the instrument has been properly set up, appropriate assay and instrument controls have been applied, and, if necessary, the instrument and method validations have been performed, operators may encounter and address instances when troubleshooting is necessary.
The most common flow cytometry challenges are high fluorescence or side scatter background, abnormal event rates, high fluorescence intensity, and low fluorescence signal. Approaches to alleviating these issues are described below.
High Particulate Background
Excessive cell handling (e.g., vortexing), improper fixation, and bacterial contamination of the cells can all increase the particulate background. In addition, if the instrument’s forward threshold is set too low, cell debris will be detected as events. Gentle cell handling, fresh reagents, and appropriate instrument settings help ensure consistent side scatter profiles.
High Fluorescence Background
High fluorescence intensity can be attributed to excessive antibody concentration, inadequate cell washing, or inadequate Fc receptor blocking. In addition, improperly high instrument PMT gain can also result in a high background. Consistent antibody concentration and cell density, adequate washing and blocking, and appropriate instrument settings will help avoid abnormally high fluorescence background.
High Event Rate
Abnormally high event rates are often attributed to high cell densities during antibody staining or in the final cell sample. Inadequate mixing and settling of the cell sample can result in high cell event rates, as can improper or inconsistent gating.
Low Event Rate
Cell clumping, low final sample cell densities, blockages in the instrument fluidics, or improper gating can often result in abnormally low event detection. Proper cleaning, maintenance, and setup of the instrument, as well as consistent staining protocols, can help achieve consistent results with sufficient sensitivity.
High Fluorescence Intensity
As in the case for high fluorescence background, high mean cell fluorescence can result from too much labeled antibody, inadequate or inconsistent cell washing, or inadequate blocking. Including detergent in the wash buffer, especially during intracellular staining, can help prevent nonspecific antibody binding.
Weak Fluorescence Intensity
Many factors can result in weak fluorescence intensity. Instrument parameters such as poor laser alignment, improper compensation, improper setup, inconsistent gain settings, and weak laser output can all negatively affect fluorescence intensity. In addition, cell physiology or reagent preparation issues, such as insufficient antibody concentration, labile or secreted target antigen, poor-quality or improperly stored reagents (resulting in fluorochrome fading), or inaccessible target antigen, can all result in a weak signal. Adequate assay development, proper instrument maintenance, and adherence to qualified protocols can all improve the fluorescence signal intensity.
Flow cytometry enables investigators to analyze cells for many different applications. Types of immunophenotypic and functional assays are increasing in number and in scope. The presence of proteins and cellular processes and detection of rare or abnormal cell populations can be studied. The reader is referred to the technical literature for application and method details.
REFERENCES
Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W. Current Protocols in Immunology. Fourth ed. Hoboken, NJ: John Wiley and Sons; 1994.
Shapiro HM. Practical Flow Cytometry. Fourth ed. Hoboken, NJ; John Wiley and Sons; 2003.
1
AlexaFluor series (InVitrogen/Molecular Probes) or the Cy series (GE Healthcare).
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Fouad Atouf, Ph.D.
Director - Biologics Biotechnology (301) 816-8365 |
(GCBA2010) General Chapters - Biological Analysis |
USP38–NF33 Page 732
Pharmacopeial Forum: Volume No. 36(5) Page 1267