



NUCLEIC ACID EXTRACTION
Introduction
The basic principles of nucleic acid amplification technology (NAT) and definitions of the various techniques are covered in Nucleic Acid-Based Techniques—General  1125
1125 . The current chapter covers general steps in the extraction and purification of nucleic acids from a variety of samples.
. The current chapter covers general steps in the extraction and purification of nucleic acids from a variety of samples.
The expanding discipline of molecular biology in pharmaceutical and biomedical research and development is characterized by the rapid discovery of new markers for disease and technologies for their detection. Nucleic acid targets are isolated from a wide variety of specimens, and the quality and quantity of the extracted target are highly affected by specimen collection, handling, and choice of extraction procedure.
The analysis of complex organisms by molecular biological techniques requires the isolation of pure, high molecular weight genomic DNA and intact full-length RNA. The application of these techniques then allows the detection, identification, and characterization of the associated organism or adventitious agent. Recently developed tests employing purified human DNA enable genetic testing for the presence, predisposition, or carrier status of inherited diseases such as cystic fibrosis, hereditary hemochromatosis, or Tay–Sachs disease, to name a few examples, or the analysis of single nucleotide polymorphisms (SNPs).
DNase and RNase are the major sources of nucleic acid instability. Although both enzymes are ubiquitous and are easily released during nucleic acid extraction, RNases are far more stable and harder to inactivate than are DNases because they generally do not require co-factors in order to function. Minute amounts of RNase are sufficient to destroy RNA, so great care should be taken to avoid inadvertently introducing these enzymes into the sample during or after the isolation procedure. If RNA is collected for the specific application of gene expression analysis, researchers should keep in mind that the sample collection process itself can alter the resulting expression profile.
Because of the ubiquity of RNases, measurement of intracellular RNA targets has lagged behind that of DNA targets in contributing to patient management and characterization of targets for pharmaceutical purposes. However, RNA represents the current status of the organism and is an important tool for correlating a phenotype with its associated genetic activity. The unstable nature of RNA has made standardization of NAT tests difficult, and false negative results can easily arise from a poorly handled sample because of target degradation rather than from the absence of disease or regulation of gene activity. Nevertheless, commercially available isolation and detection systems provide a high level of standardization and robustness, resulting in the implementation of RNA-based assays in recent years. The following sections discuss general steps in the extraction and purification of nucleic acids from a variety of samples, focusing on (1) collection, handling and storage of samples; (2) disruption of samples; (3) subsequent extraction and purification of nucleic acids; and (4) storage of purified nucleic acids.
Sample Source
The broad diversity of possible specimens requires different procedures for collection. For example, blood samples are collected in an appropriate anticoagulant- or additive-containing tube. Ethylenediaminetetraacetic acid (EDTA) and acid citrate dextrose (ACD) are the recommended anticoagulants for tests that require plasma or bone marrow aspirate (BMA) samples. When extraction from tissues is appropriate, the optimal amount of tissue is usually 1 to 2 g, depending on the type of tissue, because the amount of DNA and RNA per weight of tissue varies greatly from tissue to tissue. In general, more than 10 mg of tissue is required to obtain >10 µg of DNA or RNA. Because of the highly variable amounts and types of proteins and other contaminants present in different tissues, nucleic acid isolation protocols are tissue-specific, and a broad range of ready-to-use isolation systems are available from different manufacturers of kits for nucleic acid extraction. The tissue type also influences the stability of both DNA and RNA in specimens, and the two types of nucleic acid differ importantly with respect to sample preparation and downstream analysis. These issues are described later in the chapter.
Pre-Analytical Steps and Sample Collection
Although the genetic makeup of the organism remains mostly unchanged over time, the mRNA population represents the current status of a cell under any given set of conditions, and thus is highly dynamic. To prevent degradation of mRNA and/or to preserve the original transcription pattern of the cellular mRNA, tissue should be placed immediately on ice or snap-frozen in liquid nitrogen. However, freezing disrupts the cellular structure and releases RNases. Hence, for RNA isolation in general (mRNA, ribosomal RNA, viral RNA, etc.), thawing in an RNase-inactivating buffer is essential. A more convenient procedure employs a stabilizing agent at ambient temperature. Several reagents for different types of sample material (e.g., tissue or bacteria) are commercially available. Vanadium salts were once used to inhibit RNase activity, but they have been superseded by the use of chaotropic agents for the inhibition of RNase and stabilization of RNA. The sample can easily be collected in such reagents and stored for several days to weeks prior to RNA isolation.
For reliable gene-expression analysis, the immediate stabilization of the RNA expression pattern and of the RNA itself is an absolute prerequisite. Directly after the biological sample is harvested or extracted, changes in the gene-expression pattern occur because of specific and nonspecific RNA degradation as well as transcriptional induction. Such changes in the gene-expression pattern should be avoided for all reliable quantitative gene-expression analyses, such as biochip and array analyses and quantitative reverse transcription-polymerase chain reaction (RT-PCR).
The use of gloves while handling reagents and RNA samples is mandatory to prevent RNase contamination arising from contact with the surface of the skin or from laboratory equipment. In order to create and maintain an RNase-free environment, laboratory personnel should treat water or buffer solutions with diethylpyrocarbonate (DEPC), which inactivates RNases by covalent chemical modification. Care should be taken because DEPC is irritating to the eyes, skin, and mucous membranes and is also a suspected carcinogen. Alternatively, commercially available RNase-free solutions and reagents may be used. Commercially available RNase inhibitor proteins are also available for use in reactions but with different levels of effectiveness with respect to various RNase types. However, it should be noted that DEPC cannot be used with Tris-buffered solutions. Many scientists recommend the use of disposable vessels when working with RNA. Nondisposable glassware should be cleaned with a detergent, thoroughly rinsed, and oven baked at 240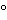 for 4 or more hours before use (autoclaving alone will not fully inactivate many RNases). Alternatively, glassware can also be treated with DEPC. Nondisposable plasticware should be thoroughly rinsed with 0.1 M sodium hydroxide and 1 mM EDTA, followed by RNase-free water. Alternatively, chloroform-resistant plasticware can be rinsed with chloroform to inactivate RNases. The use of aerosol-resistant filter tips is also important for avoiding RNase contamination. These issues are not critical for DNA, and following the rules of Good Laboratory Practice (GLP) is generally sufficient for successful isolation of DNA.
for 4 or more hours before use (autoclaving alone will not fully inactivate many RNases). Alternatively, glassware can also be treated with DEPC. Nondisposable plasticware should be thoroughly rinsed with 0.1 M sodium hydroxide and 1 mM EDTA, followed by RNase-free water. Alternatively, chloroform-resistant plasticware can be rinsed with chloroform to inactivate RNases. The use of aerosol-resistant filter tips is also important for avoiding RNase contamination. These issues are not critical for DNA, and following the rules of Good Laboratory Practice (GLP) is generally sufficient for successful isolation of DNA.
As a general precaution, staff should follow all applicable safety precautions when handling tissue or body fluids (human or other). Some of these precautions (e.g., the use of disposable gloves) also prevent contamination of the sample. Applicable guidelines and standards for the collection and processing of human-derived materials have been published by the American Association of Blood Banks, the International Conference on Harmonization, and the FDA.
Sample Disruption and Homogenization
Prior to extraction, source material is disrupted and homogenized. Disruption is the complete breakage of cell walls and plasma membranes of solid tissues and cells in order to release all DNA and RNA contained in the specimen. This is usually done using a lysis buffer that also inactivates endogenous nucleases. In addition to disrupting tissues, homogenization shears high molecular weight DNA and cellular components. During RNA isolation, scientists often must reduce the viscosity of cell lysates (caused by the presence of high molecular weight DNA molecules) prior to final isolation in order to make the subsequent extraction steps easier and more efficient. Incomplete homogenization may interfere with subsequent RNA purification steps (e.g., inefficient binding of RNA to silica membranes) and therefore result in significantly reduced yields. A typical procedure to shear high molecular weight DNA and homogenize the sample is to repeatedly pass the lysate through a small-gauge needle. However, this procedure is time-consuming and is not suitable for high throughput of samples. Better procedures to achieve complete disruption and homogenization of cells and tissue include rapid agitation in the presence of beads and lysis buffer (bead milling) or rotor–stator homogenization.
During the bead milling process, disruption and simultaneous homogenization occur by the shearing and crushing action of the beads as they collide with the cells. Disruption efficiency is influenced by the size and composition of the beads, the speed and configuration of the agitator, the ratio of buffer to beads, the disintegration time, and the amount of starting material. These parameters must be determined empirically for each application. For disruption with mortar and pestle, the samples should be frozen in liquid nitrogen and ground to a fine powder under liquid nitrogen. Standard safety precautions and the use of safety clothing to protect the skin and eyes should be employed when working with liquid nitrogen. Rotor–stator homogenizers are able to disrupt and homogenize animal and plant tissues within 5 to 90 seconds, depending on the sample. The rotor turns at very high speed, causing the sample to be disrupted by a combination of turbulence and mechanical shearing. Other alternatives are commercial spin-column homogenizers in combination with silica-membrane technology, which provide a fast and efficient way to homogenize cell and tissue lysates without cross-contamination of samples.
In order to achieve complete disruption, different sample types require different procedures. Cells from tissue culture grown as a monolayer or in suspension are easily disrupted by the addition of a lysis buffer that typically contains a mixture of an anionic detergent, a protease, and a chaotropic agent in a buffered salt solution. In contrast, nucleic acid isolation from fibrous tissues such as skeletal muscle, heart, and aorta can be difficult to disrupt because of the abundance of contractile proteins, connective tissue, and collagen. Fresh or frozen tissue samples should be cut into small pieces to aid lysis. Blood samples, including those treated to remove erythrocytes, can be efficiently lysed using a lysis buffer and a proteinase.
In general, the same procedures are applicable for extraction of DNA and RNA. For DNA isolation more gentle procedures are preferable, but during RNA isolation, cells and tissues can be disrupted using a mixer mill because there is no risk of shearing the RNA. Certain downstream applications require high molecular weight DNA, and care should be taken not to shear the DNA molecules and thus render the DNA unsuitable for further analysis.
Extraction and Purification
Although several procedures are available for nucleic acid extraction, the suitability of a procedure depends on the starting material, the type and purity of nucleic acid isolated, and possibly the downstream application. The principal procedures are described below; several commercial kits are available to accommodate different sample types and applications.
Phase Extraction—
The original technique for extraction of DNA and RNA from lysed samples is phase extraction, which involves nucleic acid extraction using a mixture of phenol and chloroform. Depending on pH and salt concentration, either DNA or RNA partitions in the aqueous phase. At neutral/basic pH, the DNA remains in the aqueous phase, and RNA remains in the organic phase or in the interphase (with the proteins). However, at acidic pH, DNA in the sample is protonated, neutralizing the charge and causing it to partition into the organic phase. RNA, which remains charged, partitions in the aqueous phase. The two phases are separated by centrifugation, and the aqueous phase is re-extracted with a mixture of phenol and chloroform, followed by extraction with chloroform to remove any residual phenol. The nucleic acid is recovered from the aqueous phase by precipitation with alcohol. For RNA, this procedure is often combined with a protease digestion, alcohol or lithium chloride precipitation, and/or cesium chloride (CsCl) density gradients. A potential problem is contamination of the recovered DNA or RNA with organic solvents that may interfere with enzymatic downstream applications or spectrometry readouts.
Cesium Chloride Density Gradient Centrifugation—
For the isolation of high molecular weight genomic DNA, CsCl density gradient centrifugation is the traditional procedure. Cells are lysed using a detergent, and the DNA is isolated from the lysate by alcohol precipitation. The DNA is then mixed with CsCl and ethidium bromide and centrifuged for several hours at a high g force (typically 100,000 × g). The DNA band, which can be visualized under UV light as a result of the intercalation of the ethidium bromide with the DNA, is collected from the centrifuge tube, extracted with isopropanol to remove the ethidium bromide, and then precipitated with ethanol to recover the DNA. This procedure allows the isolation of high-quality DNA, but it is time consuming and also a safety concern because of the high quantity of EtBr involved.
Anion-Exchange Chromatography—
An alternative procedure for the purification of high molecular weight genomic DNA is anion-exchange chromatography based on the interaction between the negatively charged phosphate groups of the nucleic acid and positively charged surface molecules on the anion-exchange resin. Binding occurs under low-salt conditions, and impurities such as RNA, cellular proteins, and metabolites are washed away using medium-salt buffers. Pure DNA is eluted with a high-salt buffer and is desalted and concentrated by alcohol precipitation. This procedure yields DNA of a purity and biological activity equivalent to two rounds of purification in CsCl gradients, but in much less time. The procedure also avoids the use of toxic substances, and it can be adapted for different scales of purification. DNA up to 150 kilobases (kb) in length may be isolated using this procedure. Several kits are available for the isolation of DNA based on anion-exchange technology, and procedures vary in processing times and the quality and size of the isolated DNA.
Silica Technology—
The current procedure of choice for most applications is based on silica technology and can be used for isolation of full-length RNA or DNA with an average size of 20 to 50 kb. However, higher molecular weight DNA exceeding 100 kb is not efficiently extracted by this technology. The procedure relies on the selective adsorption of nucleic acids to silica in the presence of high concentrations of chaotropic salts. Although both types of nucleic acid adsorb to silica, the use of specific buffers in the lysis procedure ensures that only the desired nucleic acid is adsorbed while other nucleic acids, cellular proteins, and metabolites remain in solution. The contaminants are washed away, and high-quality RNA or DNA is eluted from the silica using a low-salt buffer. The silica matrix can be used as particles in suspension, in the form of magnetic beads, or as a membrane. This technique is suitable for high throughput, and several kits and automated systems are commercially available. However, these aqueous lysis buffers (in contrast to lysis buffers based on an organic solvent such as phenol) are not ideally suited for difficult-to-lyse samples (e.g., fatty tissues). Kits designed to facilitate lysis of fatty tissues and to inhibit RNases are available. Silica-based kits provide a fast and reliable procedure for both DNA and RNA purification and are commonly used for nucleic acid extraction.
Although these procedures yield pure nucleic acids, for some applications in which even trace contaminations with either RNA or DNA may interfere, pretreatment with DNase or RNase may be necessary. Alternatively, procedures that use specific probe capture may be used. Relevant applications requiring such ultra-pure nucleic acids are discussed in Nucleic Acid-Based Techniques—Amplification 1127.
Specific Applications for Hard-to-Extract Materials
Extraction from Formalin-Fixed and Paraffin-Embedded Biopsies—
The nucleic acids in formalin-fixed paraffin embedded (FFPE) biopsies are usually heavily fragmented and chemically modified by formaldehyde. Although formaldehyde modification cannot be detected in standard quality control assays such as gel electrophoresis, formaldehyde modification does interfere with enzymatic analyses. Sufficient extraction and demodification for DNA can be achieved by prolonged digestion with protease, but this will lead to heavy fragmentation and degradation of RNA. Some isolation systems have been optimized to reverse as much formaldehyde modification as possible without further RNA degradation. Nevertheless, RNA purified from FFPE samples should not be used in downstream applications that require full-length RNA. Some applications may require modifications to allow the use of fragmented RNA (e.g., designing small amplicons for RT-PCR).
Extraction from Bacteria and Pathogens—
Although Gram-negative bacteria are relatively easy to lyse, Gram-positive bacteria or yeasts typically need an enzymatic pretreatment to remove the cell wall for efficient lysis. This methodology can be applied only to DNA isolation because the enzymatic treatment will influence the expression profile of the organism, and therefore RNA isolation requires a more rapid lysis procedure. Another factor to consider is that microorganisms normally occur against the background of a host or an environmental matrix (e.g., soil), which makes detection by polymerase chain reaction (PCR) often difficult because of inhibitory components. This means that the isolation procedure has to be carefully adapted and optimized for the specific organism and sample type. Commercial kits are available, and most are based on the use of lysozyme for the removal of cell walls.
Special Considerations for Limited Sample Amounts—
Multiple genetic testing techniques, including SNP analysis, short tandem repeat analysis, sequencing or genotyping using arrays, real-time PCR, and other procedures depend on the availability of high-quality DNA. Because human genomic DNA or samples of individual genotypes are often limited, a process to immortalize nucleic acid samples can overcome this limitation. Procedures applicable to genotyping are discussed in Nucleic Acid-Based Techniques—Genotyping 1129. Whole-genome amplification (WGA) has recently been employed to amplify limited genomic DNA from already purified DNA or directly from clinical or casework samples without any DNA purification. Two basic technologies for WGA are available and are PCR-based or rely on isothermal multiple-displacement amplification. These applications are described in more detail in Nucleic Acid-Based Techniques—Amplification 1127.
Sample Handling and Long-Term Storage
DNA is a relatively stable macromolecule, and once isolated it can be kept at 2 to 8 for at least 1 year. However, where DNA is present in very small quantities, such as in a test of residual DNA, it may be advisable to store the DNA at less than or equal to  20. Generally, DNA is stored in solution. Distilled water can be used if DNA will be used for PCR and/or endonuclease digestion within a few days after its isolation. However, Tris–EDTA at pH 7.5–8.5 is the preferred buffer for DNA storage because DNA degradation can occur in water because of the limited buffering capacity of this medium. Purified nucleic acids retain recognizable characteristics during long-term storage, provided the samples are stored as frozen solutions. The DNA solution should be stored as a primary stock solution frozen at –80. DNA can also be lyophilized and stored dry without the need for refrigeration. In some cases DNA can be stored for years on special filter papers that bind DNA and allow storage in a dried state at ambient temperature.
20. Generally, DNA is stored in solution. Distilled water can be used if DNA will be used for PCR and/or endonuclease digestion within a few days after its isolation. However, Tris–EDTA at pH 7.5–8.5 is the preferred buffer for DNA storage because DNA degradation can occur in water because of the limited buffering capacity of this medium. Purified nucleic acids retain recognizable characteristics during long-term storage, provided the samples are stored as frozen solutions. The DNA solution should be stored as a primary stock solution frozen at –80. DNA can also be lyophilized and stored dry without the need for refrigeration. In some cases DNA can be stored for years on special filter papers that bind DNA and allow storage in a dried state at ambient temperature.
The ubiquity of RNases requires extra precautions when handling RNA. Isolated RNA should be kept on ice when aliquots are pipetted. Filter tips that prevent RNase carry-over from the pipette and sterile, disposable polypropylene tubes are recommended throughout the procedure because these tubes are generally RNase-free and do not require any pretreatment to inactivate RNases. Purified RNA can be stored at –20 or –80 in water. Under these conditions no degradation is normally detectable. Unlike DNA, RNA does not benefit from basic buffer solutions during long-term storage because of its sensitivity to alkaline conditions. Generally, if nucleic acid samples are required for multiple testing, RNA and DNA samples should be frozen in multiple aliquots at –80 for subsequent analysis in order to avoid repeated freeze–thaw cycles that can lead to degradation, and also to minimize the possibility of contamination, which could result in analytical inaccuracy.
QUALITATIVE AND QUANTITATIVE EVALUATION OF NUCLEIC ACIDS
Introduction
This section describes procedures that assess the purity, integrity, and quantity of purified nucleic acids, including spectroscopic procedures, electrophoresis of nucleic acid fragments, and probe-based techniques. Detection and quantitation by amplification are discussed in Nucleic Acid-Based Techniques—Amplification 1127.
absorbance spectroscopy
The basic principles of spectroscopy are addressed in Spectrophotometry and Light-Scattering 851. For nucleic acids, absorbance is determined at 260 nm, but this procedure does not distinguish between DNA and RNA. Absorbance can also be used to estimate protein contamination in nucleic acids. Proteins maximally absorb at 280 nm, and nucleic acids maximally absorb at 260 nm. Thus the calculation of the A260/A280 ratio is used as an estimation of protein contamination in nucleic acid preparations. A ratio of 1.8 to 2.0 is considered desirable. As an example, double-stranded DNA has an extinction coefficient of 20 for 1 mg per mL of DNA at 260 nm and a coefficient of 10 at 280 nm. In contrast, for 1 mg per mL of protein, the extinction coefficients are on the order of 1 at 280 nm (depending on tyrosine and tryptophan content) and 0.57 at 260 nm. Thus a large protein contamination could exist at a 260/280 ratio of greater than 1.8 because of the lower sensitivity of protein absorbance. In addition, the change of absorbance of DNA with wavelength (DA/D ) is steep at 280 nm, and this could lead to an incorrect determination if the spectrophotometer is out of calibration. The peak at 260 nm is broad, and thus readings are less sensitive to calibration issues.
) is steep at 280 nm, and this could lead to an incorrect determination if the spectrophotometer is out of calibration. The peak at 260 nm is broad, and thus readings are less sensitive to calibration issues.
Information on contamination by nonproteinaceous materials can be provided by a scan of DNA from 220 nm to 320 nm. Pure DNA has a mostly symmetric peak around 260 nm, zero absorbance at 320 nm, and a minimum at 230 nm. Absorbance rises again from 230 nm to 220 nm. Interfering substances can co-purify with DNA and absorb in the lower UV range (around 230 nm). These substances can interfere with and lead to an overestimation of DNA content, thus showing the utility of a scan—or at least a measurement of absorbance—at 230 nm in addition to 260 nm and 280 nm. Absorbance above 300 nm can arise from other contaminants and particulate matter. Common reagents used in the isolation of DNA, particularly solvents such as phenol and alcohols if they are not completely removed, can interfere with DNA absorbance measurements. Analysts should be aware of the limitations of this type of measurement. Finally, the absorbance of DNA and the 260/280 ratio is dependent on ionic strength—a difference as large as 30% can exist. Absorbance of genomic DNA is higher, and the 260/280 ratio is lower in pure water when compared with the same DNA in a buffer or a salt solution.
For the purposes of quantitation of nucleic acids, the respective extinction coefficients for DNA and RNA are used. An absorbance of 1 in a 1-cm cuvette corresponds to 50 µg per mL of double-stranded DNA [E (specific absorption coefficient) = 0.02 (µg per mL)1 cm1]. The specific absorption coefficient for RNA at 260 nm is E = 0.025 (µg per mL)1 cm1 (absorbance of 1.0 corresponds to 40 µg per mL), and for single-stranded DNA E = 0.027 (absorbance of 1.0 corresponds to 37 µg per mL). A solution of DNA is read against a blank of the same buffer solution in which the DNA is dissolved. Ideally, readings should fall within a range of 0.1 to 1.0 absorbance for adequate linearity. Absorbance above 1.0 becomes increasingly nonlinear as the absorbance rises. The accuracy of readings below 0.1 (5 µg per mL DNA) depends on the quality and noise level of the spectrophotometer.
Fluorescence Protocols for DNA and RNA Quantitation
Cyanine dye derivatives are used for the quantitation of nucleic acids because they specifically interact with nucleic acids (DNA, RNA, and oligonucleotides) and fluoresce only upon binding. The exact mechanism of interaction is not always fully understood but may involve intercalation in double-stranded DNA and surface binding.
Measurements can be performed using a fluorometer or a plate reader. The sensitivity of fluorescence with these dyes is much higher than that of absorbance, which gives these dyes great utility when DNA concentration is low (down to 25 pg per mL). The dye must be protected from light to avoid photobleaching. Linearity is maintained over three to four orders of magnitude. Calf thymus and Lambda phage DNA are often used as calibrants to construct a standard curve. Some of these dyes have been optimized to bind double-stranded DNA or single-stranded RNA and oligonucleotides. A DNA-binding dye will also bind to single-stranded DNA and RNA but at low ionic strength, and the signal is about 10% or less than that seen with double-stranded DNA for an equivalent mass of material. Thus, this methodology is preferred for DNA measurements when no effort has been made to remove RNA from the preparation. Another fluorescent dye is available and is optimized for RNA measurements. Using two different concentrations of this dye, analysts can detect RNA in amounts as low as 1 ng per mL and as high as 1 µg per mL. The dye also fluoresces with DNA but does not display an equivalent ability to minimize binding by the use of particular conditions (e.g., with DNA and the double-strand binding dye). Quantitation may be affected by contaminating nucleic acid (e.g., DNA in an RNA preparation and vice versa). Treatment with a DNase is needed if DNA is present in the RNA preparation. Proteins are unlikely to interfere with these dyes, but some detergents as well as phenol result in loss of fluorescence. Nucleic acid extraction reagents should thus be checked for effect on subsequent fluorescent assays.
Bisbenzimide fluorochrome dyes such as (2¢-[4-hydroxyphenyl]-5-[4-methyl-1-piperazinyl]-2,5¢-bi-1H-benzimidazole) represent another option for measuring DNA. Researchers have studied the binding of these dyes to the minor groove of DNA and have found that sequences of adenine or thymine in the DNA sequence provide a minor groove dimension that binds the dyes best. Thus the fluorescent signal can show DNA sequence dependence, and the calibrant DNA should have a nucleotide composition that is similar to that of the DNA to be measured. These dyes are not as sensitive as cyanine dyes but are more sensitive than absorbance measurements. Low dye concentrations and high ionic strength are required in order for analysts to distinguish double-stranded DNA from RNA. Low ionic strength conditions are required in order to differentiate double-stranded DNA from single-stranded DNA.
Detection by Size
Agarose Gel Electrophoresis—
Agarose gel electrophoresis provides a simple and accurate procedure for separating nucleic acids by fragment size. The technique can be adapted to separate fragments over a large range of sizes and can be used in a preparative or analytical fashion. For example, gel electrophoresis can be used to verify that a product of a PCR reaction is of the correct size. DNA fragments can be retrieved from a gel slice and provide a sufficiently pure PCR product for cloning or sequencing. The general integrity of an RNA preparation can be determined by gel electrophoresis as well. The stoichiometry of the nucleic acid fragment size (in base pairs) and negative charge from the phosphate provide the basis for the separation. With the exception of plasmids, electrophoresis is generally free of DNA conformation-induced effects. Supercoiled plasmid DNA will migrate ahead of linear or open-circle/nicked plasmid, which is useful for determining the conformation of a plasmid preparation. In contrast, denaturing gels are used for RNA because of RNA's tendency to form inter- and intramolecular secondary structures.
Agarose gel electrophoresis utilizes a horizontal setup wherein the gel is cast in a box and placed on a bridge between two buffer compartments that are filled with the buffer of choice. The gel is also covered with a thin layer (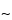 1 mm) of buffer. Although the main electrical resistance resides in the gel itself, there is sufficient charge on the nucleic acids to move fragments through the gel toward the anode. The fragments move in proportion to size, the smallest moving the fastest. The parameters that most affect electrophoresis are gel pore size, buffer concentration, and the voltage gradient. The ability to separate the fragments of choice is largely a function of the gel pore size, which depends on agarose concentration. Generally the agarose concentration is in the range of 0.5% to 1.0% for DNA fragments of <100 to 25,000 base pairs, and the higher concentration is used when it is important to distinguish the smallest fragments. Lowering the agarose concentration in the gel results in the resolution of larger fragments but also in a loss of resolution of small fragments. For the largest fragments pulsed, (reversed)-field electrophoresis is utilized.
1 mm) of buffer. Although the main electrical resistance resides in the gel itself, there is sufficient charge on the nucleic acids to move fragments through the gel toward the anode. The fragments move in proportion to size, the smallest moving the fastest. The parameters that most affect electrophoresis are gel pore size, buffer concentration, and the voltage gradient. The ability to separate the fragments of choice is largely a function of the gel pore size, which depends on agarose concentration. Generally the agarose concentration is in the range of 0.5% to 1.0% for DNA fragments of <100 to 25,000 base pairs, and the higher concentration is used when it is important to distinguish the smallest fragments. Lowering the agarose concentration in the gel results in the resolution of larger fragments but also in a loss of resolution of small fragments. For the largest fragments pulsed, (reversed)-field electrophoresis is utilized.
To achieve uniform electrophoresis, all of the agarose must be completely melted. Electrophoresis-grade agarose is dissolved in the same buffer that will be used for electrophoresis. The buffers most commonly used for DNA separations are TBE (tris-borate-EDTA) or TAE (tris-acetate-EDTA). TBE has a higher buffering capacity than TAE, but TAE should be used if the DNA is going to be retrieved from the gel. Denaturing RNA gels use MOPS buffer (40 mM MOPS, 10 mM sodium acetate, 1 mM EDTA, pH 7.0). Melting the agarose is conveniently achieved with the assistance of a microwave oven. The agarose will easily come to a boil, but this may not result in complete melting of the agarose, which may require bringing the solution to a boil several times, with intermittent mixing and holding periods, until the agarose is completely melted. Agarose particles transform from white to transparent before melting. Any partially melted agarose can be detected by swirling the flask while holding it up to the light. If the solution does not appear uniform, then it requires additional heating. The agarose is poured into the gel box after partial cooling but before setting up. Commercially available ready-to-use gels suitable for a particular application can also be used. For RNA-denaturing gels, formaldehyde is added under a fume hood to the melted agarose to a final concentration of 2.2 M or 6.7%. Before the agarose has hardened, the analyst places a comb in the gel to provide wells for the samples and size standards. Once solidified, the gel is placed in the electrophoresis box, and buffer is added until both sides are filled and there is a layer of buffer across the surface of the gel. Then 10X tracking buffer (40% sucrose with 0.25% bromphenol blue or 0.25% xylene cyanol or both) is added to each DNA sample to increase the sample density and to provide a tracking dye that is used to assess when the electrophoresis is finished. The increased density allows the sample to be transferred into the well and to remain there until it migrates into the gel during electrophoresis.
One or more lanes should be used for a DNA size standard containing fragments in the range that is relevant to the samples and agarose concentration. Size standards in various ranges are readily available. Bracketing the samples in wells between standards is useful to determine whether the electrophoresis gradient has been uniform over the width of the gel. However, in the case of eukaryotic RNA preparations, the 18S and 28S ribosomal RNAs that are co-extracted from prominent bands (corresponding to 1900 and 4700 nucleotides) can also be used as size standards. In addition, the rRNA provides information on the RNA integrity because missing or fuzzy rRNA bands indicate problems with the quality of the RNA preparation. Once the wells are filled, the cover is placed over the gel box, and the box is connected to the power supply. The indicator dye in the tracking buffer added to the samples and size standard allows the easy determination of how far the electrophoresis has proceeded. Bromophenol blue will migrate with DNA fragments of <500 base pairs, and xylene cyanol will migrate with fragments of 5000 base pairs.
The power supply is frequently run under conditions of constant voltage (1 to 10 V per cm) of gel length. Elevated voltage can cause high current, resulting in the generation of damaging heat and exhaustion of the buffer.
Pulsed-Field Electrophoresis—
This variation on agarose gel electrophoresis is used to separate a range of large DNA fragments and is most useful when resolution of 50,000 to 200,000 base-pair fragments is needed. The main difference is the addition of an alternating-field device that controls the power supply operating under constant voltage. Large fragments of DNA change conformation in order to move through the agarose pores, and the larger pieces take longer to readjust when the field is reversed and thus move more slowly than do smaller fragments. This allows resolution of fragments over the period of hours that the pulsed-field procedure operates. A commonly used ratio of forward to reverse is 3:1, and, in addition, the procedure typically calls for a stepwise increase in the unit time between reverses of the field. Electrophoresis may continue for 10 to 16 hours to avoid fluctuation in gel temperature, viscosity, and other properties that may cause artifacts.
Polyacrylamide Gel Electrophoresis (PAGE)—
The format for performing PAGE is quite different from that for agarose gel electrophoresis. The general procedure for PAGE is described in Biotechnology-Derived Articles—Polyacrylamide Gel Electrophoresis 1056. For resolution of small fragments of DNA in the 10 to 500 base-pair range, nondenaturing polyacrylamide gel electrophoresis is more suitable than agarose gel electrophoresis because separation of fragments of this size requires much smaller pore size than is achievable in agarose gels. The gel is prepared by polymerization of acrylamide monomers. The percentage of acrylamide dictates the range of fragment sizes that can be best resolved. For example, 20% acrylamide is suitable for the 10–100 base-pair range, and 5% acrylamide is useful in the 100–500 base-pair range. Commercially available ready-to-use polyacrylamide gels suitable for the particular size discrimination can also be used. The separated nucleic acids are visualized by staining with, for example, silver nitrate solution rather than with ethidium bromide or cyanine dye. However, staining with silver nitrate is laborious and time-consuming and not suitable for preparations that contain a large amount of protein, because proteins will also stain with silver nitrate.
Capillary Electrophoresis and Laser-Induced Fluorescence (CE–LIF)—
CEF has been used for many years to separate DNA fragments (for the general principles of CE, see Biotechnology-Derived Articles—Capillary Electrophoresis 1053). The procedure relies on a principle similar to that underlying agarose gel electrophoresis. CE can utilize the cross-linked buffer systems applied in gel electrophoresis, but the technique can also use polymer-containing solutions (e.g., polymethylcelluloses) that are designed to create pores that entangle proteins. These polymer solutions may be added to the capillary between injections, allowing a “fresh” gel prior to each run. In addition, capillaries can be used for more injections than are possible for polymerized gel-filled capillaries. The resolving power of the separation depends on the size of the pores, which is based on the composition of the gel. Kits are available to separate fragments into the desired size ranges. Fragment sizes outside the resolution window can possibly be separated, but the separation may not be reliable or reproducible when the gel capability is exceeded.
Fragments can be detected by a variety of mechanisms. Detection utilizing UV absorbance is possible, but the preferred and most common detection procedure is laser-induced fluorescence (LIF). Fluorescence offers improvements over UV detection in terms of selectivity and sensitivity. In addition, the detection limits for fluorescence are two to three orders of magnitude better than those for UV. Although DNA is intrinsically fluorescent, the background fluorescence and complex laser spectroscopy required preclude routine use. The most common way to label DNA is described in the section above on fluorescent protocols for RNA and DNA quantitation. This system is widely employed because of its simplicity (the dyes are added to the sample or into the reaction buffer) and effectiveness. The advantages of CE include speed of analysis, sensitivity using minimum sample volumes, and the potential for automation. These are achieved mainly by the inherent miniaturization of the gel. Automated systems allow robust analysis of the quality, quantity, and fragment size of both RNA and DNA. CE applications have been especially important for evaluating the integrity of RNA because of the instability and progressive degradation of RNA caused by ubiquitous RNases, and new technologies that compare the ratios of 28S and 18S are improving the capabilities of these procedures.
FILTER HYBRIDIZATION AND IN VITRO LABELING OF PROBES
Introduction
Hybridization techniques were used early in molecular biology to identify individual nucleic acids and to estimate the degree of similarity between species. Hybridization is widely used in the procedures described in this and other chapters to visualize and identify nucleic acid sequences (see Nucleic Acid-Based Techniques—Amplification 1127, Nucleic Acid-Based Techniques—Genotyping 1129, and Nucleic Acid-Based Techniques—Approaches for Detecting Trace Nucleic Acids (Residual DNA Testing) 1130). With the advent of restriction endonuclease digestion of DNA and electrophoretic separation by molecular mass, hybridization using labeled probes provided a way to visualize the organization of genes within a specific genome.
The hybridization techniques described are dot and slot blotting, Northern blotting, Southern blotting, in situ hybridization, and fluorescent in situ hybridization (FISH). All these techniques rely on the use of nucleic acid probes. Probes are oligonucleotides with specific DNA or RNA sequences that have been labeled with radioactive, fluorescent, chemiluminescent, chemical tags or enzymes (reporter molecules). Hybridized probes bind to complementary sequences on the target nucleic acids and are used to visualize and characterize targets, as described below.
Dot and Slot Blotting
Dot blotting is the simplest and quickest of the hybridization techniques. The nucleic acids are directly applied to a support membrane, which may be a nitrocellulose or nylon membrane, without prior separation of the nucleic acid species by agarose gel electrophoresis. The nucleic acids are spotted onto the filter using a micropipettor or an apparatus such as a dot blot or slot blot apparatus. This consists of a membrane frame with a membrane sandwiched in between the two pieces of the frame. The bottom frame plate is connected to a vacuum manifold, and the top piece of the frame has slots through which the nucleic acids are loaded. The samples are loaded under vacuum and pulled through the membrane by vacuum, with the nucleic acid binding to the membrane, and then the filter is air-dried. The nucleic acids are fixed to the filter either by heating to 80 for nitrocellulose membranes or by exposure to UV light for a predeterrmined time for nylon filters. Hybridization with a labeled probe provides confirmation of the identity of the nucleic acid but does not provide any information about the number or sizes of the species. The nucleic acid species of interest can be quantitated by spotting known concentrations of the purified nucleic acid on the filter and comparing the signal generated by the unknown samples with those of the standard preparations.
Southern Blotting
Southern blotting refers to the transfer of DNA from an agarose or polyacrylamide gel to a nitrocellulose or nylon membrane. Small, single-stranded DNA probes can then be used to visualize and identify the DNA species of interest. Southern blot analysis is based on a transfer and immobilization methodology developed in 1975, coupled with the electrophoretic separation of fragmented DNA. More specifically, the procedure typically is used to identify specific nucleic acid sequences in the context of a defined genetic topography, such as a restriction endonuclease map. The position of genes within the viral genome can be accurately mapped using a variety of restriction endonucleases in combination with Southern blot analysis. The procedure requires that DNA be obtained in sufficient quantity for analysis. Fragmented DNA is separated according to size using agarose gel electrophoresis. Double-stranded DNA fragments must be denatured before they are transferred and immobilized on a membrane by capillary action. The immobilized DNA is then cross-linked to the filter, which may be composed of nitrocellulose or nylon, as described above. However, the use of positively charged nylon membranes eliminates the need to fix the DNA to the nylon membrane. Nitrocellulose membranes are more fragile and may be probed up to 3 times with separate probes. Nylon membranes are more robust and may be probed 10 to 12 times, but they may present more background noise, particularly when they are used with chromogenic probes.
Northern Blotting
Northern blot analysis comprises a series of steps for the separation, transfer, and immobilization of RNA in a manner similar to the treatment of DNA using Southern blot analysis. Denaturation of the RNA is required to reduce secondary structure to ensure that the RNA separates in the agarose uniformly according to length. Denaturation of RNA is accomplished either prior to electrophoresis using glyoxal or dimethyl sulfoxide (DMSO) or during electrophoresis by means of gels that contain formaldehyde. Transfer is achieved in a manner identical to that used for Southern blotting. However, in the case of Northern blotting, it is unnecessary to denature the RNA prior to transfer because denaturation is accomplished before electrophoretic separation of the RNA species. The immobilized RNA is cross-linked to the membrane in a manner similar to the cross-linking of DNA.
In Situ Hybridization and Fluorescent In Situ Hybridization (FISH)
Hybridization of a nucleic acid in situ classically refers to determining the location of that nucleic acid sequence in its natural state—in tissue, in individual cells, or on a chromosome. In situ hybridization probes are designed to bind to complementary nucleic acid sequences, whether they be DNA or RNA. The purpose of these hybridization procedures is to discover where in a tissue a gene is being expressed, in which case the target is RNA, or to map a specific DNA sequence to its location on a chromosome, in which case the target is DNA.
Chromosome mapping of DNA sequences is accomplished by chemically attaching silver grains to the probe sequences and then counting the density of the grains in a metaphase chromosome spread. Although, historically, these procedures worked well, sensitivity was always an issue. The solution was to use a reporter that was more sensitive and safer than the other reporters, namely, fluorescence used in the technique of fluorescent in situ hybridization (FISH). FISH has an additional benefit in that the different colors available in fluorescence afford the ability to observe multiple hybridization events simultaneously, a feature not available with other detection systems.
Detection of DNA and RNA in Hybridization Assays Using Labeled Probes
Visualization and location of individual nucleic acid species of interest are achieved by the specific hybridization of DNA or RNA probes that are labeled for easy visualization. The filter or sample (fixed cells or tissues in the cases of in situ hybridization and FISH) is incubated with the labeled probe at an appropriate temperature and salt concentration that allows hybridization of desired stringency. This is followed by washing with buffers of varying detergent and salt concentrations and at varying temperatures in order to minimize background signal due to nonspecific hybridization. The labeling and types of probes are discussed below.
Probes can be RNA probes generated in vitro or DNA probes, either double-stranded fragments, plasmids, or single-stranded oligonucleotides containing moieties to facilitate detection of fragments that contain portions of the gene of interest. Probes can be labeled with radioactive tracers such as 32P or 35S by incorporation of a labeled nucleotide in the probe sequence or with a nonradioactive label such as biotin by incorporation of a modified base, such as adenine monophosphate linked to biotin. Radioactive probes are visualized with X-ray film placed over the blot. Biotin-labeled probes are detected with a conjugate of streptavidin–alkaline phosphatase. An enzymatic reaction is run with alkaline phosphatase and a substrate that yields an insoluble colored product at the site of the probe. Variations on nonradioactive probes utilize other modifications to the DNA and linked antibody–alkaline phosphatase, as well as chemiluminescent probes that are detected on film.
Nucleic acids can be synthesized and manipulated by either enzymatic or chemical means. These same systems can be used to modify nucleic acid structure and to introduce foreign moieties to create unique molecules that can provide an advantage to the detection of limiting viral nucleic acids against a background of host nucleic acids. The chemical synthesis of nucleic acids and their purification has become routine, and high-quality synthesis and purification are commonly achieved. Moreover, larger segments can be synthesized, and when even larger segments are required, the subsections can be designed for concatenation and ligation.
Custom synthesis of DNA oligonucleotides is readily achievable in the laboratory using commercially available reagents and equipment. Alternatively, probes can be custom ordered from numerous commercial providers. Size-exclusion procedures for purification generally are used to eliminate incomplete oligonucleotides. RNA oligonucleotides also may be chemically synthesized or generated in vitro using complementary cloned DNA fragments under the control of various prokaryotic RNA polymerase promoter sequences. The use of DNA probes is much more common, but there may be some applications in which the increased association of RNA–RNA or RNA–DNA hybrids is advantageous.
The principal procedures of labeling DNA are direct labeling using a kinase reaction to attach a labeled nucleotide to the end of each DNA strand, by incorporating labeled nucleotides into a nicked DNA by utilizing the DNA repair function of the Klenow fragment of Eschericia coli DNA polymerase I enzyme (nick translation), and by PCR. This last procedure generates a relatively higher yield of internally labeled probe because each round of thermal cycling doubles the amount of labeled probe, whereas the former procedures result in a ratio of less than one probe molecule per template molecule. The PCR procedure also is used to generate unique probes with a variety of moieties located at the termini.
NUCLEIC ACID SEQUENCING
Introduction
The first DNA sequencing procedure, described in 1977, utilized chemical cleavage to specifically introduce chain breaks in a DNA sequence (Maxam and Gilbert sequencing). The procedure proved to be of significant utility in the early years of molecular biology, but it has not been used to perform high-volume sequencing and therefore is not discussed in detail here. The majority of sequencing performed today is based on the dideoxysequencing procedure, also described in 1977 (Sanger sequencing). This procedure fundamentally changed sequencing by exploiting the enzymatic specificity of polymerases that introduce strand interruptions at specific bases. This is the most widely recognized sequencing procedure and is considered a routine assay in molecular biology laboratories. Innovations in instrumentation, sample preparation and collection, data management, data analysis, and sequence assembly have relied on this sequencing procedure as their fundamental sequence generator.
High-throughput sequencing takes all the elements of the sequencing procedures and applies them to a mass collection of sequence data, typically for larger genomes, but high-throughput sequencing certainly may be used for smaller projects as well. Obtaining the final sequence information includes all processes associated with sample preparation, sequencing, data assembly, and data finishing. The technology to achieve these individual objectives includes the instrumentation, disposables, protocols, and procedures.
Sequencing Reaction
The dideoxysequencing procedure takes advantage of specificity of the Klenow enzyme to introduce chain-terminating nucleosides, called dideoxynucleotides, intermittently during the polymerase extension process. The sequencing of each sample requires four separate reactions (one for each base). The resulting mixture of various nucleotide chain lengths is then separated on the basis of individual molecular masses. The incorporation of radioactively labeled nucleotides during the sequencing reaction permits the detection of the nucleotide chains.
Improvements in biotechnology have led to the discovery of more robust enzymes with high fidelity, improved stability, and other attributes that have led to longer reads and improved sequence fidelity. These improvements have made possible the introduction of cycle sequencing, which is now commonly used. The principle of the cycle sequencing procedure is a combination of Sanger sequencing and aspects of PCR amplification, whereby dideoxynucleotides are incorporated into the amplified DNA. Cycle sequencing leads to a higher concentration of labeled fragments covering a wider range of sizes than does Sanger sequencing, leading in turn to a higher read length.
Separation Procedures for DNA Sequencing Fragments
The previous sections of this chapter deal with the treatment of intact DNA and RNA molecules; the following sections address the challenges of separating the fragments that result from the sequencing reactions, notably slab gel sequencing and capillary electrophoresis. Subsequent sections address detection technologies and sequence integrity.
Slab Gel Sequencing
Polyacrylamide gel electrophoresis, frequently referred to as slab gel electrophoresis, was the first separation mechanism employed for the separation of DNA sequencing fragments. As described above, the electrophoretic separation of DNA fragments is driven by the size of the fragments in the reaction mixture. However, for slab gel sequencing the pore sizes are chosen so that single-base resolution for many hundreds of bases is possible. In addition to the polyacrylamide in the gel, a denaturant such as urea is frequently included to ensure denaturation of the fragments. Until the implementation of multicapillary sequencing systems, the separation power and throughput of slab gel separation mechanisms were often considered state of the art.
Capillary Electrophoresis Sequencing
As noted above, capillary electrophoresis offers significant advantages over gel-based separations. However, as with slab gel sequencing, the pore sizes are chosen so that single-base resolution for many hundreds of bases is possible. Multicapillary systems that utilize 8 to 384 capillaries are commercially available. These systems are the primary systems used for large-scale DNA sequencing, and, theoretically, they yield more than 1.1 billion base pairs of DNA sequences per year.
Detection
Radioactivity—
The first detection strategies for DNA sequencing reactions utilized radioactive isotopes such as 32P or 35S, primarily because these were practical for gel separations. The advantages are that detection is universal, low limits of detection are possible, mobility shifts are eliminated, and fidelity differences for the DNA polymerases do not occur. Disadvantages include the high disposal and safety costs, the inability to multiplex (ultimately limiting throughput), and the need for 24 to 36 hours of exposure time (i.e., no real-time detection).
Fluorescence—
Fluorescence dyes have largely replaced radioactive isotopes as detection tools during DNA sequencing, mainly because they do not have the disadvantages of radioactive probes. Because the dyes can be discriminated by means of their emission maxima, multiplexing is possible, so four sequencing reactions per sample can be replaced by a single reaction using four different labels. Thus a single lane can be used rather than the four separate lanes that were necessary with radioactive probes. Additional advantages are higher throughput and automated data collection in real time.
Mass Spectrometry—
Mass spectrometry (MS) has revolutionized the field of biochemistry and has significant potential in the area of nucleic acid sequencing. Soft-ionization techniques such as electrospray ionization and matrix-assisted laser desorption–ionization have expanded the potential application of MS to DNA sequencing. MS offers some advantages over other detection methodologies, including speed of fragment detection (signal acquisition is in the range of microseconds versus hours for conventional approaches) and accuracy (e.g., the molecular mass of each fragment can be determined with a high degree of accuracy). The Sanger procedure makes use of mass differences of the fragments generated as part of the polymerization reaction. MS is sufficiently precise to resolve fragment sizes that differ by only one base pair. Unfortunately, the sensitivity of MS detection suffers as fragment length increases, and the 100-base-pair barrier has yet to be crossed.
More recently, other sequencing technologies have emerged that are based on massively parallel sequencing techniques that attempt to achieve low-cost sequencing. These techniques are based, for example, on solid-phase sequencing or they make use of highly parallel and miniaturized pyrosequencing, which is described in Nucleic Acid-Based Techniques—Genotyping 1129.
Sequence Integrity
A prerequisite for automated data collection and interpretation is that the data must be of good quality, which means minimizing human intervention and allowing the system to make base identifications following detection steps. It is a critical step to ensure accurate base identification by minimally sequencing both strands of the DNA several times. In addition, other tactics may be employed, such as using primers at different sequence positions, which can improve the accuracy of the developed consensus sequence. This task can be facilitated by the use of specialized software packages that are commercially available. More recent technology developments have produced alternative sequencing platforms that are more amenable to large-scale sequencing projects. These techniques include array-based platforms on which short stretches of target are sequenced on a chip that supplies raw data to sophisticated computational programs that reconstruct the sequence. Other sequencing approaches have been developed for the rapid sequencing of short nucleic acid sequences such as oligonucleotides of short PCR products. These technologies include MS-based and pyrosequencing platforms, the latter of which is described in Nucleic Acid-Based Techniques—Genotyping 1129.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Anita Y. Szajek, Ph.D.
Principal Scientific Liaison (301) 816-8325 |
(GCBA2010) General Chapters - Biological Analysis |
USP38–NF33 Page 1237
Pharmacopeial Forum: Volume No. 33(5) Page 990