



SCOPE
This chapter provides guidance for the packaging and repackaging of single-unit containers, and for the use and application of unit-of-use packaging. Although the chapter is intended for use by drug manufacturers, repackagers, and pharmacists, the information in the chapter may also be useful for suppliers of packages and packaging components. For the definition of specific types of packaging, see Packaging and Storage Requirements  659
659 .
.
SINGLE-UNIT CONTAINER
Single-unit containers that package a prescription drug to be dispensed directly to the patient are required to be child-resistant. Single-unit packaging intended for institutional or hospital use may or may not be required to be child-resistant. Single-unit containers that are child-resistant include supported blisters, such as separate, peel, push, and tear notch, and enclosed or in-card blisters, such as pull tabs and slide packs.
PACKAGING MATERIALS
Materials used to manufacture single-unit packaging containers include glass and plastic. Glass used as a primary packaging component should meet the requirements of Containers—Glass 660. Plastic materials as a primary packaging component should meet the requirements of Containers—Plastics 661. The test for moisture permeability may be carried out as described in general test chapter Containers—Performance Testing 671.
PACKAGING CLOSURE TYPES
Reclosables and nonreclosables may be used for solid, semisolid, and liquid dosage forms. Both must be packaged in compliance with the 16 CFR 1700.15 standards. The Poison Prevention Packaging Act (PPPA) of 1970 requires in certain cases the use of special packaging—child-resistant and senior-friendly. Child-resistant packaging protects children from serious injury or illness resulting from ingesting or handling hazardous products including drugs.
Because drugs packaged in unit-of-use packaging are intended to be dispensed to the consumer without repackaging by the pharmacist, the manufacturer or repackager is responsible for the special packaging of PPPA-regulated substances in unit-of-use containers (16 CFR 1701.1).
Reclosables
Reclosables are containers with suitable closures that may incorporate tamper evidence and child-resistance capabilities. Reclosables may be used for glass or plastic containers.
Nonreclosables
Nonreclosables are containers with closures that are nonreclosable, such as blisters, sachets, strips, and other single-unit containers. Nonreclosables may include packs such as cold-formed foil blisters, foil strip packs, and PVC/Aclar® combining multilayer materials that are thermo-formed or cold-formed foil blisters. Nonreclosables may be child resistant depending on the intended use and place of use. Household nonreclosables are subject to the PPPA as defined in 16 CFR 1700.14. However, because of some unit-dose designs, not all unit-dose packages comply with the PPPA.
UNIT-OF-USE
Unit-of-use packaging, when provided by the manufacturer, offers some of the following attractive advantages. (1) A dosage form can be dispensed to a patient in the manufacturer’s original container, a practice that recognizes that the suitability of the container has been established on the basis of the manufacturer's stability studies. (2) The counting and repackaging of dosage units in the pharmacy is eliminated, thereby reducing the possibility of human error. (3) The pharmacist is able to affix the label for the patient onto the unit-of-use package and is free to use the manufacturer's expiration date as the beyond-use date. (4) The number of dosage units in a single unit-of-use package may be determined on a case-by-case basis. (5) Patient compliance is improved. (6) The unit-of use package can protect against counterfeiting because traceability of product is ensured through bar coding techniques and National Drug Code (NDC) numbers.
Unit-of-use packaging, when provided by repackagers, offers the same attractive advantages as those offered by the manufacturer. However, unit-of-use repackagers should conform to all requirements as presented in Good Repackaging Practices 1178. There are a number of reasons why repackagers produce unit-of-use packaging, for example, (1) requests from institutions, (2) better inventory control, (3) reduced dispensing times, and (4) variations in some drug therapies.
The packaging of a unit-of-use system may be a multiple-unit or single-unit container. A unit-of-use system may contain a drug product in a liquid, semisolid, or solid dosage form (see also FDA Guidance for Industry, Container Closure Systems for Packaging Human Drugs and Biologics). [Note—The terms “unit-of-use package” and “unit-of-use container” may be used interchangeably. ]
Unit-of-Use Labeling
The unit-of-use containers are labeled to include expiration dates, the manufacturer’s lot number, the NDC designation, and bar codes as provided in the Labeling section of General Notices and Requirements, Preservation, Packaging, Storage, and Labeling and in Good Repackaging Practices 1178. Some of the advantages of having bar codes on the label include reduced medication errors, improved inventory control, and improved access to medication identity. The labeling covers information placed in the container by the manufacturer (see General Notices and Requirements). Acceptable labeling can range from full labeling, such as that for multiple-unit containers, to abbreviated labeling when the container is too small to include all of the text. Full labeling may also be provided on the carton if it is not present on the immediate container.
Unit-of-Use—Repackaging and Reprocessing
Unit-of-use containers are reprocessed or repackaged as instructed by the manufacturer. A unit-of-use package that is a blister package may not be reprocessed by a pharmacist once it has been deblistered from a unit-dose container (see General Notices and Requirements for application of the appropriate beyond-use date for a multiple-unit or unit-dose container). Deblistering is the process of removing medication from a blister-type container. However, under current Good Manufacturing Practices (cGMPs) and tight quality controls, the manufacturer or contract repackager may repackage and reprocess unit-of-use containers.
Information from Manufacturers
The manufacturer should provide appropriate stability information that can be used to determine appropriate labeling, storage, and shipping statements that will properly inform patients and practitioners. The manufacturer may make other assurances based on product information on packaging and distribution arrangements. In the event that a product is not to be repackaged, the manufacturer may so state in the labeling. The manufacturer also includes labeling and information necessary for optimal handling by the practitioner and the patient. The labeling and information should be bar coded to eliminate medication error and promote medication traceability.
Responsibility of the Dispenser—Labeling
The labeling on a unit-of-use container also includes a label added at the dispensing stage by the pharmacist. Prior to dispensing the unit-of-use package, the dispenser shall add label(s) that provide the following information:
- The name of the patient;
- The name and strength of the drug product, the directions for use as prescribed by a doctor or health-care provider, and the name of the prescriber; and
- Any storage instruction, beyond-use date, and other information as deemed appropriate by federal and state laws.
In the pharmacy setting, pharmacists are encouraged to use bar codes, in conjunction with computerized prescription orders, to confirm that the right drug is being dispensed to the right patient. Bar coding would minimize errors and create an opportunity for medication traceability and accountability.
Quality Control of Packaging System
The packaging system shall meet the general considerations for system suitability, protection, safety, and performance characteristics as described in FDA Guidance for Industry, Container Closure Systems for Packaging Human Drugs and Biologics, and in Containers—Glass 660, Containers—Plastics 661, and Containers—Performance Testing 671.
REPACKAGING A SINGLE SOLID ORAL DRUG PRODUCT INTO A UNIT-DOSE CONTAINER
Repackaging of solid oral drug products, such as tablets and capsules, into unit-dose configurations is common practice both for the pharmacy that is dispensing drugs pursuant to a prescription and for the pharmaceutical repackaging firm. The following section contains minimum standards to be used as a guideline for repackaging practices.
Repackaging preparations into unit-dose configurations is an important aspect of pharmaceutical care and of optimization of patient compliance. For purposes of this chapter, there are two types of repackaging: the first involves pharmacies that dispense prescription drugs, and the second concerns commercial pharmaceutical repackaging firms.
Nomenclature and Definitions
Dispenser:
A dispenser is a licensed or registered practitioner who is legally responsible for providing the patient with a preparation that is in compliance with a prescription or a medication order and contains a specific patient label. In addition, dispensers may prepare limited quantities in anticipation of a prescription or medication order from a physician. Dispensers are governed by the board of pharmacy of the individual state.
Package:
The term “package” is synonymous with the term “container”. See Packaging and Storage Requirements 659.
Pharmacy:
A pharmacy is an establishment that is legally responsible for providing the patient with a drug preparation with a specific patient label, in compliance with a prescription or a medication order. The terms “dispenser” and “pharmacy” are used interchangeably.
Repackaging:
Repackaging is the act of removing a preparation from its original primary container and placing it into another primary container, usually of smaller size.
Repackager:
A repackager is an establishment that repackages drugs and sends them to a second location in anticipation of a need. Repackaging firms repackage preparations for distribution (e.g., for resale to distributors, hospitals, or other pharmacies), a function that is beyond the regular practice of a pharmacy. Distribution is not patient specific in that there are no prescriptions. Unlike dispensers, repackaging firms are required to register with the FDA and to comply with the Current Good Manufacturing Practice regulations in 21 CFR 210 and 211.
Materials
Blister packages offer a wide array of designs both in functionality and in appearance. Various packaging materials are used to create blisters that are tailored to provide optimum performance. The blister container consists of two components: the blister, which is the formed cavity that holds the product, and the lid stock, which is the material that seals to the blister, as shown below.
+'&img=../../images/v38332/c1146-f1-usp33.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Schematic Presentation of a Typical Blister Pack
Because of the variety of blister films available, film selection should be based upon the degree of protection required. The choice of lid stock depends on how the blister is to be used, but generally the lid stock is made of aluminum foil. The material used to form the cavity is typically a plastic, which can be designed to protect the dosage form from moisture. There are widely varying degrees of moisture protection now available. For purposes of this general chapter, they are referred to as nominal, medium, high, and extreme moisture barrier properties.
Polyvinyl chloride:
The most commonly used blister material is polyvinyl chloride (PVC). This material, which provides a nominal or zero barrier to moisture, is used when the product does not require effective moisture protection. PVC is available in a range of gauges and can be made opaque or can be tinted with pigments to block out specific light wavelengths.
The thickness of the PVC used is determined by the depth and size of the cavity to be formed. Because the plastic thins during the blister-forming process, care should be taken to ensure that the finished blister provides sufficient protection from light (if required) and that it is strong enough to adequately protect the dosage form. Common gauges of PVC used in the pharmaceutical industry range from 7.5 to 15 mil (0.0075 to 0.015 inch).
Barrier films:
Many drug preparations are extremely sensitive to moisture and therefore require high barrier films. Several materials may be used to provide moisture protection. Barrier films commonly used in the pharmaceutical industry are described below.
PVC/PCTFE laminations—
Polychlorotrifluoroethylene (PCTFE) film1 is a thermoplastic film made from polychlorotrifluoroethylene fluoropolymer. The PCTFE film is laminated to the PVC by an adhesive layer between the PVC and the PCTFE film (duplex structure),
 or by a layer of polyethylene (PE) between the PVC–adhesive and the PCTFE–adhesive layers (triplex structure).
or by a layer of polyethylene (PE) between the PVC–adhesive and the PCTFE–adhesive layers (triplex structure).
 By using various gauges of PCTFE film, medium to extreme moisture barriers can be obtained.
By using various gauges of PCTFE film, medium to extreme moisture barriers can be obtained.
+'&img=../../images/v38332/c1146-f4-usp33.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Duplex Structure
+'&img=../../images/v38332/c1146-f3-usp33.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Triplex Structure
PVC/PVdC laminations—
PVC/PVdC is a film in which the PVC is coated with an emulsion of polyvinylidene chloride (PVdC), as shown in the duplex structure pictured below.

Duplex Structure
The PVdC layer is specified in g/m2 and can be constructed to provide medium to high barrier protection. The coating weights commonly used in the pharmaceutical industry are 40, 60, and 90 g/m2, and the film is offered with or without a middle layer of polyethylene (PE), as shown in the triplex structure below. The polyethylene is used with heavier coating weights, such as 60 and 90 g/m2, to improve the thermoforming characteristics of the blister cavity.
+'&img=../../images/v38332/c1146-f5-usp33.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Triplex Structure
Polypropylene—
Because of its morphology, polypropylene (PP) serves as a good moisture barrier, its spherulitic structure creating an arduous path for water molecules to traverse. Although not commonly used as a pharmaceutical blister film in the United States, PP provides an economical alternative to medium barrier materials and is used in Europe as an alternative to PVC.
Cold form foil—
This material is used for products that are extremely hygroscopic or light sensitive. It is an extreme moisture barrier and consists of three layers: PVC, aluminum foil, and nylon.

+'&img=../../images/v38332/c1146-f6-usp33.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Cold Form Foil
Lid stock:
The lid stock is sealed to the molded blister as described above. Different designs of lid stocks are available, and selection of a particular design depends on how the package will be used. Standard designs—peelable, child-resistant peelable, and push-through—are described below. The primary component of lid stock is typically aluminum, and its gauge varies from 18–25 µm (0.0078–0.001 inch). The side of the aluminum foil laminate in contact with the product provides the heat-sealable layer that forms the seal to the blister material. The heat-seal coating should be capable of forming an adequate seal with the blister film to which it is intended to seal. The materials used in the makeup of the heat-seal layer meet the requirements of 21 CFR 175 and 177.
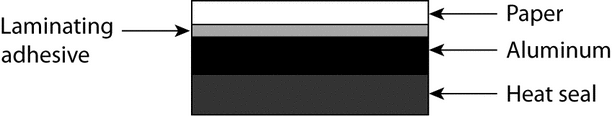
Peelable—
Peelable foil, commonly used in an institutional setting, consists of several layers, as shown below, and can be peeled away from the blister. [Note—For child-resistant peelable foil, a layer of polyester with the appropriate adhesives would be added. ] With the peelable-foil lid stock, which is used in conjunction with blister tooling, a three-step process is required to open the blister.

+'&img=../../images/v38332/c1146-f7-usp33.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Peelable Foil Construction
First, the blister cavity must be separated from the rest of the blister card. Next, the paper and polyester layers are pulled back from an unsealed area. Finally, the product is pushed through the remaining aluminum foil. It is important to note that use of this type of foil structure helps make the package more child resistant. However, if child-resistant packaging is required, the package design should be tested in accordance with the protocol described in 16 CFR 1700, the Poison Prevention Packaging Act.
+'&img=../../images/v38332/c1146-f8-usp33.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Child-Resistant Foil
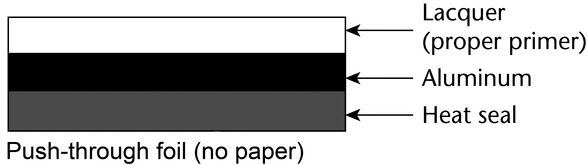
Push-through—
There are two commonly used types of push-through foil: one with a paper outer layer separated from the aluminum by a layer of adhesive, and one without paper. The paper outer layer serves as an aesthetic and makes it possible to print on the back of the blister.

+'&img=../../images/v38332/g-11464-usp33.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
+'&img=../../images/v38332/g-11463-usp33.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Other package styles:
Other types of packages used for unit-dose packaging of solid dosage forms are strip packs, pouches, and sachets.

+'&img=../../images/v38332/g-11462-usp33.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
PROCESS
Unit-dose packages can be formed and sealed in a variety of ways. Larger scale repackagers may use thermoformers that accomplish these functions in-line, while smaller repackagers may purchase preformed blister material. This section begins with an overview of the process involved in thermoforming a blister, the fundamental process that also applies to other unit-dose package types such as pouches. The overview is not intended to be all-encompassing, but it highlights the major operations along with their critical parameters.
Thermoforming a blister unit-dose package:
The complete thermoforming process consists of four basic stations where the following operations occur: forming, filling, sealing, and finishing. Thermoforming requires the use of heat and air in forming the blister. The lid stock material is sealed to the blister cavity material for a defined time (the stroke of the machine) at the point where the heat plate closes on the two materials.
Forming station—
Prior to entering the forming station, the blister material passes through a heating unit where the blister material is heated uniformly in stages to ensure proper formation. Because different plastics have different softening points, careful attention must be paid to determining the proper temperature of the heating station, which often has multiple temperature zones. The temperature, based on the blister material used and on the speed at which that material travels through the heating station, is a critical parameter for optimal performance. At the forming station, the blister material is heated to the point where the plastic softens sufficiently to allow the cavity to be formed. The blister material is drawn from a reel-mounted roll (referred to as the web) and pulled through the machine. A splicing table is located at the reel unwind to provide room for a second roll of blister material to be readily available for splicing and resumption of the packaging process. An unwind device may be installed to aid in moving the blister material from the roll as adjusted for a specific index.
Once the blister material is properly heated, compressed air is generally used to form the blister cavity. Upper and lower forming dies close on the blister material as air is introduced, forming a blister that corresponds to the size of the cavity. A plug assist may be necessary, depending on the material and size of the cavity. The plug assist ensures a uniform thinning of the blister material to optimize the protective characteristics of the formed material. Once the blister material is formed into the desired blister configuration, it is advanced to the filling station.
Filling station—
The product is loaded into the blister cavity at this station. An automated filling device may be used, or the cavities may be hand filled. The critical parameter at this station is proper filling of the formed blisters.
Sealing station—
At this station, the lid stock is sealed to the filled blister cavity, using heat and pressure for a defined dwell time. The critical parameters to be considered at this station are temperature, pressure, and dwell time.
The lid stock material is staged on a roll above the blister cavity and may be preprinted or printed online. Lot numbers and expiration dates may be applied at this point. Preprinted lid stock materials will require a print registration system to control the position of the printing relative to the blister cavity. The critical parameters at this part of the station include legible and correct labeling.
Finishing station—
The finishing station encompasses all other steps in the packaging process, including embossing, perforation, and cutting. Embossing involves application of a lot number and expiration date to the package. Steel type is used to emboss information on the edges of the blister package. One of the critical parameters at this station is package integrity. It is important that the embossing, perforation, and cutting processes do not compromise the blister, lid, or seal. The quality of the embossing is another critical parameter in the process. The embossing must be legible and correct, and must include all required information.
Pouch unit-dose packages:
The pouch process is also a form, fill, and seal operation, but it does not provide a defined, formed cavity as does the thermoforming process. Although the equipment used to form pouch unit-dose packages may function differently from that described for thermoforming a blister, the main operations (form, fill, and seal) and critical parameters at those stations are quite similar. [Note—See the critical parameters defined in the section on thermoforming. ]
The strip-pack process involves the drug product being dosed into a three-sided, formed pouch. Once filled with the drug, the machine seals the pouch, forming a strip of sealed unit-dose pouches. The basic flow of the process begins with the drug situated above the pouch material. One roll of strip-pack material is used to form the pouch. This is accomplished by moving the material over a device that forces the material to fold into two equal sides. The sides and bottom are sealed prior to dosing. The strip pack may be cut later during the equipment processing or roll continuously and be manually cut. Temperature and dwell time are the main critical factors for this equipment.
Preformed unit-dose packages:
Preformed containers are sealed either by heat or by adhesion. Heat sealers may be manual units requiring hand pressure application or automated units that provide a more controlled pressure for sealing.
Heat sealing may be accomplished through the use of manual tabletop equipment, which is generally operated at a set pressure. Critical parameters with these devices are pressure and temperature control because undesirable variation in these parameters may yield inadequate seals.
Critical parameters:
In order to ensure that the finished container performs as intended, qualification of critical parameters should be determined. Typically, validation of a packaging line consists of qualification of the installation, operation, and performance of a packaging system.
Installation qualification—
Equipment should be installed and found to be in proper working condition prior to use.
Operational qualification—
Operational qualification should be performed to establish that the equipment operates within the manufacturer’s specified ranges. Incoming utilities for the equipment, such as air, electricity, etc., should be monitored and checked periodically.
Performance qualification—
Performance qualification should be done to establish that the equipment performs properly with the required materials to produce a container that functions as intended. The critical parameters include forming temperature and pressure, sealing temperature and pressure, and dwell time at the seal station. Qualified ranges should be readily available in a reference source for the setup of equipment. Re-evaluation may be necessary with changes to equipment, materials, or process.
In-process inspections:
Strict controls covering the packaging and labeling processes should be in place. The final container should be evaluated for performance in each of the stations previously described. Specifically, the formed container should be inspected visually to ensure that it is properly formed. Evaluation of the filling station should include a check to ensure that the unit dose is properly filled (i.e., that the correct product is present). The sealing station should be evaluated to ensure that a proper seal has been made and that the moisture permeation specifications of the sealed container have been met. A visual examination of the package should be performed to ensure that the final steps of the packaging process are acceptable.
Repackagers and dispensers should use a standard inspection plan to verify the adequacy of the package. A visual inspection should be performed to verify that the correct product is in the proper packaging materials with correct labeling. Seal integrity should be evaluated, using vacuum testing,2 helium testing, tear testing, and other testing methods suitable to establish whether seal integrity is maintained.
PERFORMANCE
The primary purpose of the unit-dose package used in the packaging of a drug preparation is to ensure that, until its intended expiration date, there is adequate protection from the environment as the dosage form is distributed and stored. It is also essential that the materials used do not interact with the dosage form.
When determining what type of package to use in the repackaging operation, consideration must be given to the dosage form’s sensitivities (if any) to the storage and distribution environments (e.g., temperature, light, and moisture).
The properties of the finished container are defined by the materials used in constructing the unit-dose container, and by the process used to form and seal the container. As discussed in Materials, there is a wide variety of commercially available film structures that provide unit-dose containers with a range of moisture and light protection. Suppliers of these materials typically provide quantitative data, obtained from well-established test methods, to highlight the protective properties of their material. These data are based on flat sheets of the film, not on the formed container.
It is critical to understand that once the film is formed, protective properties change because the overall thickness of the film decreases as the blister cavity is formed. Usually the change is a decrease, especially in the case of barrier properties. However, the extent of change will vary with the type of film structure used and is also highly dependent on the container-forming process used (see Process). Further, a suboptimal seal on the formed container will decrease the protective properties of the container. Insufficient temperature, time, or pressure during a heat-seal operation may enable the passage of moisture or oxygen through the seal area over time, which may have an effect on the dosage form. In addition, if the seal area is designed with insufficient surface area, the same problem may occur. To ensure a good seal, a minimum sealing distance of 3 mm from the edge of the blister cavity to the nearest edge or perforation is recommended. Therefore, it is important to measure the performance of the formed and sealed container rather than the performance of the flat sheet.
Moisture is a critical factor in preparation integrity. Containers—Performance Testing 671 describes how to determine and classify moisture permeation rates. If the manufacturer’s labeling includes “Protect From Moisture,” the repackager shall utilize a high barrier film.
If light protection is required for a drug preparation, the repackager should follow the requirements for light transmission established under Containers—Performance Testing 671. Again, this testing should be conducted on the formed container, because the light-protective properties of the film are compromised once the film is thinned during the forming process. It is recommended that these tests, in conjunction with any guidance provided by the manufacturer, be considered appropriate for any container–closure system used in repackaging a drug preparation.
BEYOND-USE-DATE
In the absence of stability data for the drug product in the repackaged container, the beyond-use dating period is one year or the time remaining until the expiration date, whichever is shorter. If current stability data are available for the drug product in the repackaged container, the length of time established by the stability study may be used to establish the beyond-use date, but must not exceed the manufacturer’s expiration date.
As stated in the General Notices and Requirements, the dispenser must maintain the facility where the dosage forms are packaged and stored at a temperature such that the mean kinetic temperature is not greater than 25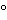 . The plastic material used in packaging the dosage forms must afford better protection than polyvinyl chloride, which does not provide adequate protection against moisture permeation. Records must be kept of the temperature of the facility where the dosage forms are stored, and of the plastic materials used in packaging.
. The plastic material used in packaging the dosage forms must afford better protection than polyvinyl chloride, which does not provide adequate protection against moisture permeation. Records must be kept of the temperature of the facility where the dosage forms are stored, and of the plastic materials used in packaging.
MINIMUM REQUIREMENTS
The previous sections serve as a general introduction to repackaging by providing a basic understanding of materials selection, the form-fill-seal process, and the importance of performance of the sealed container. In this section, certain minimum requirements for repackaging, which must be met, are described in more detail.
Personnel:
Each person with responsibility for the repackaging of a preparation shall have the education, training, and experience, or any combination thereof, to perform assigned functions in a manner such that the safety, identity, strength, quality, purity, potency, and pharmaceutical elegance of the drug dosage form are retained. Training should be documented.
Personnel engaged in the repackaging of a preparation shall wear clean clothing appropriate for the duties or processes performed.
Facility:
The repackaging facility may require areas of low relative humidity, and temperature conditions should meet controlled room temperature requirements specified in Packaging and Storage Requirements 659.
Equipment:
Equipment used in the repackaging of a preparation shall be of appropriate design and suitably located to facilitate operations for its intended use. Its design should allow for cleaning to preclude cross-contamination as well as for maintenance to be performed. Equipment shall be constructed so that those surfaces that contact components or a preparation are not reactive, additive, or absorptive. Any substances required for operation, such as lubricants or coolants, shall not come into contact with components or a preparation.
Equipment and utensils shall be cleaned, maintained, and sanitized at appropriate intervals to prevent malfunctions or contamination. Preventive maintenance should be performed at appropriate intervals in accordance with the equipment manufacturer’s recommendation. Any instruments used to monitor critical parameters should be calibrated on a defined schedule.
Process:
Steps should be taken to determine the critical process parameters (e.g., seal temperature, dwell time) in operating the equipment. Set points for these parameters should be documented and procedures established to ensure that they are adhered to each time the equipment is operated.
Labeling:
The labeling requirements for a commercial repackager and a pharmacist are different. For example, the commercial repackager must comply with 21 CFR 201.1, but the pharmacist or dispenser does not have to comply with this requirement. If stability data are unavailable, the dispenser shall repackage only an amount of stock sufficient for a limited time and shall include product name and strength, lot number, manufacturer, and appropriate beyond-use date on the label. When quantities are repackaged in advance of immediate needs, each preparation must bear an identifying label, and the dispenser is required to maintain suitable repackaging records showing the name of the manufacturer, lot number, expiration date, date of repackaging, and designation of persons responsible for repackaging and for checking. The repackager or dispenser will use documented controls to prevent labeling errors.
Materials:
The repackager or dispenser shall place an appropriate beyond-use date on the label and package in appropriate materials. Materials used by the repackager shall not be reactive, additive, or absorptive, and must meet the requirements described in 21 CFR 175 and 177.
Storage:
The dispenser shall rotate and monitor stock closely to ensure that the dispensing of preparations is on a first-in-first-out (FIFO) basis. The repackager or dispenser shall store preparations under required environmental conditions (e.g., controlled room temperature with a mean kinetic temperature not higher than 25).
Drug product:
The repackager or dispenser shall examine preparations for evidence of instability such as change in color or odor, and shall exercise professional judgment as to the acceptability of a package.
Complaints:
The repackager or dispenser will maintain written procedures describing the handling of written and oral complaints regarding a drug product and will ensure that complaints are investigated and appropriately resolved.
Returned goods:
Policies and procedures relating to returned goods should be developed to ensure proper handling.
Reprocessing:
Reprocessing of repackaged unit-dose containers (i.e., removing medication from one unit-dose container and placing it into another unit-dose container) shall not be done. However, reprocessing of the secondary package (e.g., removing the blister card from the cardboard carrier and placing the blister card into another cardboard carrier) is allowed provided the original beyond-use date is maintained, and provided the integrity of the blister is ensured.
Special considerations:
If a product is known to be oxygen sensitive or if it exhibits extreme moisture or light sensitivity (e.g., cold form foil), it shall not be repackaged. If a product is refrigerated, it shall not be repackaged unless proper environmental conditions and suitable materials are available. Certain drug products (such as oncologic agents, hormones, or penicillin derivatives) require special handling because they are considered very potent or toxic, and because transfer of any portion of these products to another product could have deleterious effects.
REPACKAGING NONSTERILE SOLID AND LIQUID DOSAGE FORMS INTO SINGLE-UNIT CONTAINERS AND UNIT-DOSE CONTAINERS
The following guidance is intended for those engaged in pharmaceutical dispensing, and does not apply to commercial dispensing. An official dosage form is required to bear on its label an expiration date assigned for the particular formulation and package of the article. This date limits the time during which the product may be dispensed or used. Because the expiration date stated on the original manufacturer’s container–closure system has been determined for the drug in that particular system and is not intended to apply to a product that has been repackaged in a different container, repackaged drugs dispensed pursuant to a prescription are exempt from using the expiration date from the original manufacturer’s package. However, under no circumstance should the repackaged pharmaceutical preparation’s expiration date exceed the original manufacturer’s expiration date. It is necessary, therefore, that other precautions be taken by the dispenser to preserve the strength, quality, and purity of drugs that are repackaged for ultimate distribution or sale to patients.
The following guidelines and requirements are applicable where official dosage forms are repackaged into single-unit or unit-dose containers or mnemonic packs for dispensing pursuant to prescription.
Labeling:
It is the responsibility of the dispenser to place a suitable expiration date on the label, taking into account the nature of the drug repackaged, any packaging and expiration dating information in the manufacturer’s product labeling, the characteristics of the containers, and the storage conditions to which the article may be subjected. Repackaged dosage forms must bear on their labels expiration dates as determined from information in the product labeling (see General Notices and Requirements, Preservation, Packaging, Storage, and Labeling). Each single-unit or unit-dose container bears a separate label, unless the device holding the unit-dose form does not allow for the removal or separation of the intact single-unit or unit-dose container therefrom.
Storage:
Store the repackaged article in a humidity-controlled environment and at the temperature specified in the individual monograph or in the product labeling. For further directions, see Packaging and Storage Requirements 659.
A refrigerator or freezer shall not be considered to be a humidity-controlled environment. Drugs that are to be stored at a cold temperature in a refrigerator or freezer must be protected during storage in the refrigerator or freezer. An outer container may be necessary for such protection; it is recommended that the drug monograph be referenced for storage.
Reprocessing:
Reprocessing of repackaged unit-dose containers (i.e., removing a dosage unit from one unit-dose container and placing it in another unit-dose container) shall not be done. However, reprocessing of the secondary package (e.g., removing the blister card from the cardboard carrier and placing the blister card into another cardboard carrier) is allowed provided that the original expiration date is maintained.
CUSTOMIZED PATIENT MEDICATION PACKAGES
In lieu of dispensing two or more prescribed drug products in separate containers, a pharmacist may, with the consent of the patient, the patient’s caregiver, or a prescriber, provide a customized patient medication package (patient med pak).3
A patient med pak, i.e., a package prepared by a pharmacist for a specific patient, comprises a series of containers and contains two or more prescribed solid oral dosage forms. The patient med pak is so designed, or each container is so labeled, as to indicate the day and time, or period of time, that the container contents are to be taken.
It is the responsibility of the dispenser to instruct the patient or caregiver on the use of the patient med pak.
Label:
The patient med pak shall bear a label stating the following:
- The name of the patient;
- A serial number for the patient med pak itself and a separate identifying serial number for each of the prescription orders for each of the drug products contained therein;
- The name, strength, physical description or identification, and total quantity of each drug product contained therein;
- The directions for use and cautionary statements, if any, contained in the prescription order for each drug product therein;
- Any storage instructions or cautionary statements required by the official compendia;
- The name of the prescriber of each drug product;
- The date of preparation of the patient med pak and the beyond-use date or period of time assigned to the patient med pak (such beyond-use date or period of time shall be not longer than the shortest recommended beyond-use date for any dosage form included therein or not longer than 60 days from the date of preparation of the patient med pak, and shall not exceed the shortest expiration date on the original manufacturer’s bulk containers for the dosage forms included therein); alternatively, the package label shall state the date of the prescription(s) or the date of preparation of the patient med pak, provided the package is accompanied by a record indicating the start date and the beyond-use date;
- The name, address, and telephone number of the dispenser (and the dispenser’s registration number where necessary); and
- Any other information, statements, or warnings required for any of the drug products contained therein.
If the patient med pak allows for the removal or separation of the intact containers therefrom, each individual container shall bear a label identifying each of the drug products contained therein.
Labeling:
The patient med pak shall be accompanied by a patient package insert, in the event that any medication therein is required to be dispensed with such insert as accompanying labeling. Alternatively, such required information may be incorporated into a single, overall educational insert provided by the pharmacist for the total patient med pak.
Packaging:
In the absence of more stringent packaging requirements for any of the drug products contained therein, each container of the patient med pak shall comply with the moisture permeation requirements for a Class B single-unit or unit-dose container (see Containers—Performance Testing 671). Each container shall be either nonreclosable or so designed as to show evidence of having been opened.
Guidelines:
It is the responsibility of the dispenser, when preparing a patient med pak, to take into account any applicable compendial requirements or guidelines and the physical and chemical compatibility of the dosage forms placed within each container, as well as any therapeutic incompatibilities that may attend the simultaneous administration of the medications. In this regard, pharmacists are encouraged to report to USP headquarters any observed or reported incompatibilities. Once a medication has been placed in a patient med pak with another solid dosage form, it may not be returned to stock, redistributed, or resold if unused.
Recordkeeping:
In addition to any individual prescription filing requirements, a record of each patient med pak shall be made and filed. Each record shall contain, as a minimum:
- The name and address of the patient;
- The serial number of the prescription order for each drug product contained therein;
- The name of the manufacturer or labeler and lot number for each drug product contained therein;
- Information identifying or describing the design, characteristics, or specifications of the patient med pak sufficient to allow subsequent preparation of an identical patient med pak for the patient;
- The date of preparation of the patient med pak and the beyond-use date that was assigned;
- Any special labeling instructions; and
- The name or initials of the pharmacist who prepared the patient med pak.
1
PCTFE film is available from Allied Signal (as Aclar®) and from other sources.
2
Vacuum testing consists of placing samples from the packaging operation into a jar filled with water. A lid is placed over the samples to fully immerse them in the water. A container lid is applied to create a seal effective enough to create approximately 25 cm of vacuum. The vacuum pump is set, and the samples are tested for approximately 1 minute, removed from the water, wiped down, and opened to determine whether the inside of the unit-dose cavity or pouch is wet. This process should be adjusted until it is under control, and additional testing may be performed to ensure that the seal integrity is consistently acceptable. Wetness indicates a defective seal and therefore the potential for the drug to degrade when exposed to the atmosphere. Defective packages must be removed from further use.
3
It should be noticed that for patient med paks there is no special exemption from the requirements of the Poison Prevention Packaging Act. Thus, the patient med pak, if it does not meet child-resistant standards, shall be placed in an outer package that does comply, or the necessary consent of the purchaser or physician to dispense in a container not intended to be child-resistant shall be obtained.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Desmond G. Hunt, Ph.D.
Senior Scientific Liaison (301) 816-8341 |
(GCPS2010) General Chapters - Packaging Storage and Distribution |
USP38–NF33 Page 1269
Pharmacopeial Forum: Volume No. 37(6)