



INTRODUCTION
An overview of vaccines for human use is presented in Vaccines for Human Use—General Considerations  1235
1235 . Bacterial vaccines can be derived from whole cells, either killed or attenuated in their ability to cause disease, or from some component(s) of the intact cell that are important for virulence or damage to the host. Another subset of bacterial vaccines, derived from toxins, is the toxoids. Bacterial vaccine products can be mixtures of components from different species, from different strains or different serotypes of the same species, or from different components from cells of the same species.
. Bacterial vaccines can be derived from whole cells, either killed or attenuated in their ability to cause disease, or from some component(s) of the intact cell that are important for virulence or damage to the host. Another subset of bacterial vaccines, derived from toxins, is the toxoids. Bacterial vaccine products can be mixtures of components from different species, from different strains or different serotypes of the same species, or from different components from cells of the same species.
The simplest bacterial vaccines consist of the purified cell-surface capsular polysaccharides (CPS) from organisms such as Salmonella enterica serovar Typhi, various meningococcal serogroups, or pneumococcal serotypes that cause meningitis, otitis media, acute respiratory infections, and pneumonia. Although the typhoid vaccine consists of a single polysaccharide, the meningococcal vaccines contain as many as four serogroup-specific CPS, and the pneumococcal vaccine contains 23 serotypes.
The immunological response to meningococcal and pneumococcal polysaccharides, and to the capsular polysaccharide from Haemophilus influenzae type b (Hib), is improved by covalent attachment of the CPS or an oligosaccharide derived from it to a suitable carrier protein. The immunological response to these glycoconjugate vaccines is elicited via immunologic pathways different from those induced by purified polysaccharides, creates a T-cell–dependent response, and establishes immunological memory. The carrier proteins are typically bacterial toxoids or bacterial outer membrane protein vesicles but may also be from other sources. For these products, anti-CPS antibodies appear to be sufficient to protect against disease, although the glycoconjugate vaccines also may reduce carriage of the organisms in the nasopharynx. Due to the complexity of their manufacturing processes, glycoconjugate vaccine products tend to contain fewer serotype or serogroup components than do the related purified polysaccharide vaccines.
Many bacterial pathogens, including those that cause diphtheria and tetanus, produce toxins that kill tissue. Immunological neutralization of these toxins is sufficient to prevent disease. These subunit vaccines consist of chemically detoxified toxins (toxoids) purified from culture supernatant and are capable of eliciting neutralizing antibodies against the native toxin.Other types of purified subunit and purification processes may be developed.
Although earlier pertussis vaccines consisted of myriad chemically inactivated whole-cell and toxin components, current acellular products contain various combinations of specific purified proteins, sometimes toxoided (e.g., fimbriae and other cell-surface protein components). Compared to older products, these vaccines apparently produce protection by a different mode of action but have a lower incidence of adverse events. A combination of diphtheria and tetanus toxoids and an acellular pertussis vaccine form the core components of many polyvalent pediatric and adult combination vaccines. To these may also be added an Hib glycoconjugate, hepatitis B, and/or inactivated poliovirus immunogens.
Live attenuated bacterial vaccines are currently limited to Bacillus Calmette-Guérin (BCG), which protects against tuberculosis when administered through the skin, and the S. typhi Ty21a construct, which is an oral vaccine against typhoid fever.
The immune response against these bacterial polysaccharide and protein antigens can be increased by inclusion of adjuvants. The primary adjuvant licensed in the United States is based on aluminum salts such as aluminum hydroxide and aluminum phosphate, although development and characterization of new adjuvants is an active area of research.
RAW MATERIALS
Raw materials can directly affect the identity, strength, purity, and quality of bacterial vaccines. A consistent manufacturing process critically depends on use of consistent raw materials (e.g., during seed banking, fermentation, harvest, purification, and formulation; see Vaccines for Human Use—General Considerations 1235). Raw materials for bacterial growth media typically consist of both well-defined chemical entities (e.g., amino acids, carbohydrates, vitamins, minerals) and more complex components (e.g., protein hydrolysates, yeast extracts, peptones). Manufacturers should consider the source of each of these raw materials to ensure that they come from reliable vendors who adhere to cGMP quality standards and can assure a long-term supply. Manufacturers should communicate with raw material vendors in order to avoid any changes in the sourcing or manufacture of components and to avoid supply shortages. Without such communications, the consistency of the fermentation process and the supply of the vaccine can be adversely affected. Consistent raw materials are particularly critical for more complex fermentation components such as yeast extract or peptones for which changes may be difficult to detect but are likely to have a direct effect on fermentation.
Accurate records of the composition and source of the culture medium used in seed banking and routine fermentation should be maintained and also document release criteria for raw materials or components. Manufacturers should determine if any of their raw materials are derived from animal origin. If additives from animal sources are added to the culture medium, they should be certified to be free from contaminants and adventitious agents such as those that cause bovine spongiform encephalopathy or transmissible spongiform encephalopathy. Vendors/manufacturers should provide information about the identity and source of additives and should test for adventitious agents. Use of antibiotics should be minimal or should be avoided to ensure that no unwanted antibiotics are included in the drug product, unless they are intentionally used in manufacturing (e.g., as selective markers).
As manufacturers scale up fermentation to pilot production (i.e., within tenfold of final manufacturing scale), they also should ensure, to the extent possible, the availability of multiple sources for all raw materials. This will ensure that supply or business instabilities at one vendor do not become the limiting factor in vaccine manufacture.
CELL BANKS
Source and History
The source of cells used in cell banks should be documented. The original isolate should include, when possible, the age, sex, and species of the donor; the donor’s medical history; and, if available, culture history including methods used for the isolation of the substrate bacteria.
The source of cells from which the strain was derived is to be stated, and relevant references from the scientific literature should be cited. The source should generate a sufficient amount of antigen(s) to meet the medical need. Information obtained directly from the source laboratory is preferred. When this is not available, literature references can be used to provide bacterial classification (i.e., genus, species, and strain designation) and specific phenotypic and/or genotypic trait. For microbial-expression systems such as E. coli or S. pneumoniae, the manufacturer should describe the method used to prepare the DNA coding for the protein, including the cell and origin of the source nucleic acid. All propagations carried out with the original isolate should be documented and should include, as applicable, the method used for subculture, any use of animal-derived material, record of subcultivations, and storage conditions. Constituents of the culture medium must be described, in particular, materials of human or animal origin such as serum, enzymes, hydrolysates, or other living cells.
For microbial-expression systems, the steps in the assembly of the expression construct must be described in detail. This description should include the source and function of the component parts of the expression construct (e.g., origins of replication, antibiotic resistance genes, promoters, enhancers, and whether or not the protein is synthesized as a fusion protein). Manufacturers should provide restriction endonuclease digestion maps that illustrate the sites used in preparing the expression construct and sites used in identification of DNA fragments.
A complete nucleotide sequence analysis of the expression construct’s coding region for the protein of interest should be performed. The sequence analysis should be provided and should include a complete annotation designating all of the important sequence features. The copy number and physical state of the expression construct should be determined.
Cell Bank Lineage and Genealogy
A flow chart can be used to demonstrate the preparation of the cell bank lineage from the original source, through preliminary cell banks (or process development cell banks, as applicable), to the Master Cell Bank (MCB) and production Working Cell Banks (WCB).
Manufacturers should describe their strategy for providing a continued supply of cells from their cell bank(s), including the lot size and anticipated use rate of the cell bank(s) for production, the expected intervals between generation of new cell bank(s), and the criteria for qualification of cell bank(s). If multiple WCBs were used for clinical trials, process validation, or commercial supplies, flow charts can help illustrate the common source (i.e., MCB) from which the WCB were derived.
Once an MCB is produced, a cell bank system should be generated to prevent unwanted drift that might ensue from repeated subcultures or multiple generations. The system should ensure that an adequate supply of equivalent cells exists over the entire life span of the product. Ordinarily, the cell bank system consists of two tiers: an MCB and a series of WCB derived from the MCB. When additional tiers of WCBs are prepared, manufacturers should clearly identify the generation that will be used for WCB.
Cell Bank Manufacture
Generally, the MCB is made from a preliminary cell bank derived from the original source or directly from an initial clone. Manufacturers generally prepare cells for banking by expanding cultures in a progressively greater number of vessels or in larger vessels until a pool of cells is obtained. If manufacturers use more than one vessel, they can ensure the uniform composition of the contents by combining the cells from all of the culture vessels.
For microbial-expression systems, a single host cell that contains the expression construct is propagated to generate the MCB. Manufacturers should document the cell cloning history and method of transferring the expression construct into the host cell. They also should completely describe methods and criteria used to amplify the expression construct and to select the cell clone for production.
The process for WCB used in clinical trials and for commercial supply should be similar to the MCB process. A WCB is derived from one or more containers of the MCB and is typically used to directly provide cells for the manufacturing process. Additional WCB are generated from the MCB as needed.
Preferably the MCB and WCB should be prepared in a similar manner, but the MCB and WCB may differ in certain respects (e.g., culture components and culture conditions). Similarly, the culture conditions used to prepare the MCB and WCB may differ from those used for the production process or between clinical trial materials or commercial supply. The preparation procedures for all cell culture processes must be described, and details of process changes must be documented. Comparability of product quality must be demonstrated when process changes occur between WCBs.
Cell banks should be made under cGMP because they are expected to last for the lifetime of the product. The facility should be operated to minimize the chance of microbial contamination and have in place procedures to prevent cross-contamination with other materials. Critical equipment used in the preparation of cell banks should be qualified. Manufacturers should establish the cell bank in a suitably controlled environment to protect both the cell bank and personnel handling it. During the establishment of the cell bank, no other living infectious material (e.g., viruses, cell lines, or cell strains) can be handled simultaneously in the same area.
Cell Bank Validation
The cell banking process should be considered a unit operation and should be validated. The process begins with the MCB vial and the cell bank process validated for preparing WCB. The suitability of WCB for intended use should be further demonstrated by the consistency and quality of successive product batches. Qualified banks should be used for process validation of fermentation, drug substance, etc. If this is not possible, then manufacturers should perform a small-scale demonstration of the appropriateness of the cell bank. The basic principles of process validation apply, including use of validated analytical methods and stability evaluation.
Manufacturers should describe the methods used to preserve cell banks, including the cryoprotectant and media used. Storage containers (e.g., vials, ampules, and other appropriate vessels) and closure systems should be described. Container–closure systems should incorporate materials and designs that withstand storage and retrieval without breakage or leakage and are physically and chemically compatible with the stored material.
Cell Bank Testing
A newly prepared cell bank (MCB or WCB) should be evaluated by a series of appropriate release and characterization tests on an aliquot of the cell bank or on cultures derived from it, as appropriate. The amount of testing required for an MCB may influence that required for subsequent WCBs, and the extent of testing both may influence the testing needed for production cell cultures. Manufacturers should evaluate all cell banks, including bacterial cultures or recombinant bacterial expression systems, for identity, culture purity, and viability. Additionally, manufacturers should evaluate the genetic stability and consistent productivity of all cell lines.
To confirm identity, manufacturers should perform appropriate tests to determine that the banked cells are what they are represented to be. Either phenotypic or genotypic characteristics can be used in identity testing to classify bacterial strains to species level, and when applicable, supplementary serological tests can be performed. For most microbial cells and transfected cells, analysis of growth on selective media is usually adequate to confirm host cell identity. Where a variety of strains can be used, biological characterization methods such as phage typing should be considered as supplementary tests. Expression of the desired product is also considered adequate to confirm the identity of the microbial expression system.
It must also be demonstrated that cell banks are biologically pure (i.e., free from adventitious microbial agents). Testing for adventitious agents should include tests for bacteria, fungi, mycoplasmas and viruses, as applicable.
Additionally, all cell banks should be tested to confirm the viability of the cells. Viable cell counts or growth tests should be performed to demonstrate that the cell culture has sufficient viability and is suitable for its subsequent intended use.
Evaluation of genetic stability and persistence of productivity is a reflection of how many doublings the cells can tolerate without compromising their genetic integrity (e.g., plasmid retention) and productivity (e.g., mass of product per cell). Such testing is critical to ensure that the cell line performs reliably in the full course of the production process from the initial MCB stage through the longest production intended. As part of this evaluation, manufacturers should document the number of passages from the original source, the number of subcultivations from the original source to the MCB, from the MCB to the WCB, and from the WCB to the final bulk. The earliest and latest culture states (e.g., MCB and end production) should be evaluated to ensure that the desired characteristics persist. Such a demonstration of cell line stability is commonly performed once for each product marketing application.
Characterization tests may be useful for demonstrating that the cell bank is composed of cells with the intended phenotypic/genotypic characteristics. Such tests can include cellular and colony morphology (i.e., use of selective and/or differential media), biochemical profiles (enzymatic activity or substrate utilization), immunological identity, characteristic growth, and antibiotic susceptibility.
Additionally, for recombinant bacterial expression cell lines (e.g., E. coli) molecular characterization testing can include DNA sequencing of the target gene sequence along with the flanking regions, expression construct retention, and plasmid copy number. Analysis of the expression construct at the nucleic acid level should be performed with consideration that this verifies only the coding sequence of a recombinant gene. Restriction endonuclease mapping or other suitable techniques should be used to analyze the expression construct for insertions or deletions and for the number of integration sites. For extrachromosomal expression systems, the percent of host cells that retain the expression construct should be determined under selected and nonselected growth conditions. For cells with chromosomal copies of the expression construct, the nucleotide sequence encoding the product could be verified by recloning and sequencing of chromosomal copies.
Much of this testing should be conducted on the MCB if possible, which will preclude the need to repeat much of the testing on each WCB or production lot, although sometimes redundant testing (on both MCB and WCB) may be desirable.
Limited identity testing is generally performed on each WCB if extensive identity testing was performed on the MCB. For recombinant products, the identity of the WCB should be assessed by restriction endonuclease mapping of the expression construct for copy number and for insertions or deletions. In addition, where appropriate, the WCB should be identified by phenotypic characterization (e.g., auxotrophy, antibiotic resistance).
For each lot of WCB derived from the MCB, manufacturers should routinely test for contaminants that may have been introduced from the culture medium during preparation. Purity tests like those performed on the MCB to test for adventitious agents may be performed on the WCB.
Characteristics of the recombinant protein product can also be applied (see below) as another means of defining the ultimate output of the cell line.
In the event that a new MCB is needed, the testing performed on a new MCB should be the same as that performed on the original MCB unless justified. If a new MCB is to be generated by expression construct transfer into host cells followed by clonal selection, then acceptance criteria for both the new clone and the protein produced by the clone should be described and justified.
Cell Bank Storage
In both MCB and WCB of the same product, similar containers (such as cryovials) are generally used and are treated identically during storage.
The location, identity, and detailed inventory of individual ampules of cells should be thoroughly documented with procedures that allow the cell bank containers to be traced. Labeling should clearly indicate the biological name of the components, unique container number, lot or batch number if applicable, and the type of bank (such as MCB or WCB). The label must withstand storage and retrieval without loss of integrity or information.
Cell banks should be established, stored, and used in a way that minimizes the risk of contamination or cross-contamination by other cell types that may be present in storage. Once issued, banked materials cannot be returned to the controlled storage area. Access to banked material must be controlled by a strict inventory-control system with limited access by authorized individuals only.
Bacterial cell banks should be stored in either the liquid or vapor phase of liquid nitrogen or in mechanical freezers (generally 
 60
60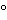 ) . Storage conditions (generally 60) may be acceptable when supported by data that demonstrate that a minimum level of cell viability is maintained and is adequate for production use. Storage temperature and other critical storage conditions should be maintained within validated limits. Temperatures must be continuously monitored and recorded, preferably on an alarm system. Shipping containers used to transport cryopreserved cell banks to offsite storage facilities or manufacturing facilities must be validated, and shipping qualification must be performed before use.
) . Storage conditions (generally 60) may be acceptable when supported by data that demonstrate that a minimum level of cell viability is maintained and is adequate for production use. Storage temperature and other critical storage conditions should be maintained within validated limits. Temperatures must be continuously monitored and recorded, preferably on an alarm system. Shipping containers used to transport cryopreserved cell banks to offsite storage facilities or manufacturing facilities must be validated, and shipping qualification must be performed before use.
Because of more frequent usage of WCBs and to protect the MCB, the WCB should be stored separately from the MCB. Cell banks may also be stored in two or more widely separate areas within the production facility, as well as at a distant site in order to avoid loss of the cell bank (e.g., caused by equipment malfunctions or disaster at the site). When stored in different locations, the cell banks must be stored under the same conditions.
As part of a disaster recovery plan, the manufacturer should document the steps and timeline needed to restart production of new cell banks and/or contingency plans for continued manufacturing production.
Storage Stability
MCB and WCB should be placed in a stability program. Evidence for banked cell stability under defined storage conditions usually is generated during production of clinical trial material or commercial material from the banked cells. Data from the determination of cell viability when the preserved cells are reconstituted for production of clinical trial supplies can verify that the revived cells have survived the preservation process. Data from the preparation of clinical materials are used to demonstrate that the revived cells can be used to prepare the desired product.
Enough MCB material for the lifetime of the product should be put on stability (enough WCB should be put on stability to support the lifetime of the WCB). This can be a large volume because the product lifetime can be quite long (e.g., 50 years). During the preparation of the MCB, the lot size should be large enough to allow adequate inventory to support the lifetime of the stability study as well as production for the life of the product. Time points for such a long-term study might include 0, 6, and 12 months, and then perhaps every 1 to 3 years thereafter. Typically, no expiration dating is used for cell banks because stability studies are used to confirm the suitability of the material. Greater reliance is placed on the successful (and typical) culture of the cells themselves. The proposed monitoring should be documented in pre-approved protocols. The time points can be reduced (e.g., increase the time between time points) if data indicate stability. In addition, time points can be added if sufficient material is available and the data suggest that more monitoring is needed. The stability plan depends on the use rate in manufacturing.
FERMENTATION
Production of the drug substance for a bacterial vaccine requires a fermentation process that is consistent and sufficiently productive to support commercial production. The approach to achieving this has become fairly standardized and provides a relatively high probability of success for early batch production to support a development program. It is still a significant challenge to achieve sufficient productivity to support commercial manufacture of a licensed product. Directly following any fermentation process is the harvest process, which serves as a transition step between biomass expansion and downstream process steps. For purposes of this chapter, harvest will be considered as an extension of the fermentation process.
Fermentation Starting Materials: Cell Inoculum
The cell inoculum for the fermentation process is the single most important component for establishing a reproducible fermentation process. In early development before finalizing fermentation conditions, manufacturers typically must generate an interim source of this inoculum, a Process Development Cell Bank (PDCB). The origin of the PDCB should be a clonal isolate of the original transfected or isolated strain that demonstrates suitable growth properties and produces the antigen of interest in sufficient quantity and quality for the intended purpose. The use of a clonal isolate ensures that the genetic starting point for each batch is the same and that subtle variations in process conditions will not inadvertently allow one population versus another to dominate the culture. That is, the PDCB is used for fermentation development to ensure that variations in the fermentation conditions can be interpreted without the overlay of competition between populations of transfectants.
Initial development of the fermentation process, preferably with the PDCB, typically precedes production of the MCB and WCB. Best practice is to derive these cell banks from the same clonal isolate as the PDCB in order to reduce the need for a second cycle of fermentation development when the WCB is deployed. Substitution of a WCB for the PDCB at the final stages of fermentation development is common practice, but care must be taken to constrain such experiments to optimization of the fermentation process. More detail is found in the cell banks section above.
Fermentation Hardware
The biomass production process typically begins with a small-volume inoculum in an initial fermentation volume that is 20- to 100-fold larger than the initial inoculum volume. This initial passage is often followed by one or more intermediate fermentations that expand the production volume by 20- to 100-fold at each step until the production fermentation volume (typically 500–3000 L) is reached. Routine manufacture at these scales requires well-controlled fermentation conditions and physical facilities that meet the economic and cGMP needs for a successful product.
Bacterial fermentations have traditionally been carried out in glass, glass-lined, or passivated stainless steel fermenters that comply with cGMP requirements, particularly when using large fermenters (e.g., those with >1000 L working volume) because of containment issues with such large volumes of liquid. Traditional fermentation systems require hard-piped control systems that meet the need for clean-in-place and steam-in-place capability. The bioburden and complexity of the facility are increased if the fermentation operations must accommodate multiple product lines as well.
Smaller fermentation batches are increasingly performed in disposable bioreactors such as single-use bags with completely disposable product contact surfaces, including sensors and probes. These systems are becoming readily available, are less expensive, and are more flexible than fixed equipment and meet the needs of the competitive business and evolving cGMP expectations and requirements. A note of caution is warranted, though, because this disposable technology can lead to changes in the material of product contact surfaces. Such changes then require re-evaluation and sometimes revalidation of the manufacturing process for late-stage development and commercial products. Thus the reduced cleaning burden may bring an increase in the need for extractability and leachability studies.
Harvest Hardware
Harvest of the fermentation product can focus on recovery of either the wet cell mass from which the product will be extracted or the fermentation broth from which the product will be directly purified. In the former case, centrifugal separation is typically employed. Production-scale centrifuges can be either closed operations with a fixed volume of input and manual recovery of the pellet or continuous-flow operations that automatically eject and recover the clarified supernatant and/or the accumulated pellet. Although centrifuges are efficient in harvesting a fermentation product, shear forces can have significant effects on the product stream (e.g., lysed cells, sheared molecules in solution). Alternatively, and particularly when the product is secreted into the solution rather than retained in the cells, membrane filtration systems may be used to clarify the product stream for subsequent purification. Tangential-flow and depth filtration systems can be effective means of recovering soluble product with less concern about shear forces.
In all cases, monitoring the processing of the fermentation output and solids removal from the liquid matrix can be simple but effective means to monitor process consistency and comparability. Off-line tools such as high performance liquid chromatography (HPLC), sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), or Western blot analyses can track product integrity issues such as aggregation or proteolysis.
Process Development
A productive, robust fermentation process is the result of careful consideration of a broad collection of variables, considered alone as well as in conjunction with product design and downstream processing. Fermentation process variables include chemical inputs (e.g., carbon sources, minerals, vitamins, trace elements, antifoam, and gases), physical inputs (e.g., temperature, mixing, and pH), and biological processes (e.g., nutrient utilization rates, metabolite levels, and inductors [quantity/type/addition duration]).
Product Requirements
A critical consideration in process development is anticipating how much of the product will be needed. Too little product caused by operating at too small a scale imposes supply constraints and often postlicensure urgency to scale up the process. In contrast, too much product results in excess inventory, expiring lots, infrequent manufacturing (itself a problem), and generally poor economics. A clear market evaluation is needed before one designs the manufacturing process or commits to a process for scale-up or -down.
Process Design
If one has a reasonably defined production need and an initial estimate of product yield, one can extrapolate the scale of the fermentation from the volume (yield), production frequency, and the expected productivity. Commercial fermentation of bacterial cultures is routinely carried out in volumes as large as 3000 L, but larger volumes are also in use. A few large lots per year can be advantageous for a very robust process but may be limited by downstream process capabilities and/or the stability of the fermentation product as a production intermediate. An additional consideration may be the difficulty in generating enough lots to ensure that the fermentation process is indeed robust. Failure of a large lot carries important financial and inventory risks.
A large number of lots can impose logistical problems if turn-around time is too tight or coordination of downstream events becomes too complex. Logistics includes quality control testing, which depends on the number rather than the size of lots. Production that involves a large number of smaller lots can also require blending of multiple intermediate lots in order to produce a final drug product lot. This can cause challenges if product-related problems occur and may entail root-cause investigations. In general, appropriate fermentation sizing results in a process that has a turn-around time of less than a week, that can be accommodated with one or a few purification runs, and that results in one to several fills of final product after each purification cycle.
Early Development Considerations
During early development of a biological product, the most important fermentation considerations are an appropriate, well-defined MCB and a fermentation process that is reasonably productive, reproducible, and scalable. The latter is often underestimated when one considers the physical, chemical, and biological control of the process as process volumes change by orders of magnitude.
The mechanics of the fermentation process are an important consideration. Fermentations are typically studied in shake flask experiments or even microscale reactors that can readily accommodate many experiments conducted in parallel. Although this is attractive for initial identification of process conditions, the ultimate culture vessel should be a controlled fermenter where growth conditions can be controlled and monitored in a more rigorous and complete manner. Manufacturers should begin work in small-scale fermenters as early as possible to ensure that robust, controlled experiments can be run to refine the initial fermentation conditions.
Fermentation harvest processes also should be scalable. Although it is possible to scale centrifugation conditions, it is a challenge to maintain equivalent centrifugal conditions, particularly in a flow-through mode. Filtration processes can usually be scaled more predictably provided the membrane manufacturer is anticipating the needs of the process development scientist.
When manufacturers define a process, they should evaluate its robustness by purposeful deviations such as changes in sources of raw materials and time and temperature limits of unit operations. Such evaluations better define the rationale for setting process limits and for knowing which are most critical to the success of the manufacturing process.
Process Monitoring
On the basis of early development process characterization data, manufacturers should be able to identify key analytical measures that, if applied to all lots, can either verify the correct progression of the process or serve as a sentinel to determine whether a specific batch is showing signs of deviating from the typical profile. In the absence of such data, an aberrant process may go unnoticed or may not be detected until testing of a process intermediate shows either an out-of-trend or out-of-specification result.
For a fermentation process, many critical variables (e.g., optical density, pH, and specific nutrient levels) can be measured online and in real time to potentially allow intervention to bring a given process back into normal range or at least to identify the point in the process at which the deviation occurred. Such data can be valuable in identifying potential process improvements. Conversely, in the absence of such data troubleshooting can be a challenging and protracted process.
Scale-up
Just as early development requires a focus on small-scale operations, scale-up becomes essential at some point to ensure that sufficiently large lots can be made to meet program needs. As these needs become increasingly complex, larger lots are essential to ensure that multiple experiments and observations can be tied to the same lot of product, which in turn is critical to understanding critical process and product variables. If proper process engineering considerations were taken into account at the smaller production scale, scale-up can usually be done in increments of ten-fold in volume with reasonable expectation that significant process performance or product changes will not be seen. This approach may require adjustments at an intermediate scale if the initial fermentation was based on too small a volume or if the final production scale is very large. Again, process monitoring data can be very helpful in evaluating the success of the scaled-up process.
If clinical development studies are performed at less than full manufacturing scale, as they usually are, manufacturers will be obliged to relate the comparability of the process performance and the product characteristics at the different scales. Analytical data can be compelling, but in their absence or in the presence of differences, manufacturers must demonstrate that scale-related differences are not clinically significant. However, the use of comparability protocols for scale changes will have to be approved by the local regulatory authority. In order to avoid fixing something that is not broken, analysts must take care to isolate differences caused by fermentation scale-up from changes caused by harvest or purification scale-up. One way to accomplish this is to compare process intermediates obtained, as possible, during the fermentation and harvest processes. As an example, online monitoring of fermentation conditions such as pH or glucose level can be used to demonstrate similarity during the time course of the fermentation. Similarly, measurements at the end of the fermentation process (e.g., final cell density, cell viability) and intermediate measurements during harvest (e.g., turbidity of clarified broth, wet cell mass in the pellet) provide useful information for evaluating the similarity or differences during scale-up.
PURIFICATION
A general overview of purification for bacterial derived vaccines is presented in USP Vaccines for Human Use—General Considerations 1235. In addition to a description of critical processing equipment, reagents, and processing steps, manufacturers should provide the rationale for the purification process chosen for component antigens recovered from the crude harvest. As with the other processes, analysts should consider the source of all raw materials and ensure that they come from reliable vendors who adhere to cGMP and can ensure a long-term supply. The cGMPs will apply to late-stage clinical supplies and commercial materials. The removal of nonproduct-related impurities (e.g., processing reagents, endotoxin, contaminating cell proteins or nucleic acids, and other residual contaminants) should be verified.
The drug substance can be one of several types of compounds: e.g., polysaccharides (wild type or modified), proteins (wild type, mutant, toxoids, or recombinant), or products of conjugation of polysaccharides and proteins, or products of conjugation of peptides and proteins.
To define and control purification processes for drug substance and drug products, the manufacturer should establish targets for process parameters and tolerances for all critical process steps including yields, activity, and purity to ensure efficacy, safety, and consistency of the final product. Requirements for pooling, if applicable, should be established. The requirements and conditions for storage of intermediates, bulks, and final containers must be established by an official stability program. The use, reuse, regeneration, and cleaning of all drug product/drug substance contact equipment (e.g., filters, chromatographic columns and resins, tanks, and process lines) should be validated. In addition, extractable/leachable studies should be performed for all product contact equipment (e.g., disposable bag systems, chromatographic column resins, and process lines).
Polysaccharide Purification
The purification steps for polysaccharides depend on the phenotype (e.g., gram negative or positive), polysaccharide presentation (e.g., membrane bound or excreted within the supernatant), and the chemical nature of the polysaccharide itself [e.g., idealized backbone linkages (glycosidic bond, phosphodiester bond), overall charge, types of charge groups, and types of side group modifications (O-acetyl, uronic acid, sialic acid, N-acetyl, pyruvate, or O-methyl)]. Polysaccharide harvest methods determine clarification and downstream purification requirements. Clarification methods depend on whether it is necessary to perform cell lysis or only to separate cell-free broth from cellular debris. Harvest techniques include centrifugation, depth filtration, tangential-flow filtration, microfiltration, sizing filtration, or a combination of techniques. The culture may be inactivated or residual contaminants removed by selective precipitation (e.g., protein denaturation) using heat or chemical treatments (e.g., salts, detergents, enzymes, or phenol). This may require cold-storage settling before clarification.
Methods for postclarification polysaccharide precipitation are used both for isolation and purification. Fractional precipitation methods are based on overall charge or cationic binding affinities of the polysaccharide (e.g., alcoholic precipitation, ion-exchange chromatography). Agents such as cationic detergents, salts, and solvents can be used to differentially precipitate charged species from uncharged molecules. Precipitation can be performed in stepwise fashion to remove residual impurities from polysaccharide or by a series of precipitations to achieve the desired purity. Polysaccharide may be contained in either the precipitate or supernatant depending on the charge and nature of the polysaccharide. Precipitation is followed by isolation steps such as centrifugation and/or filtration during which either the precipitate is discarded or resuspended in a secondary precipitating agent until the polysaccharide is recovered. Extraction with solvents such as phenol is sometimes used to remove impurities.
An alternative and additional approach to selective precipitation methods is the use of chromatographic methods. Ion-exchange chromatography (e.g., DEAE Sepharose), hydrophobic-interaction chromatography (HIC), and gel-permeation chromatography separately and in combination have been used successfully to purify polysaccharides. At neutral pH the charge on acidic polysaccharides can be used on anion exchangers to separate acidic polysaccharides from impurities. Ion exchangers can also be used to purify neutral polysaccharides in flow-through mode, binding impurities while neutral polysaccharides flow through the column. HIC can also be used to bind impurities while the polysaccharide passes in the flow-through fraction. If there are no base-labile groups, the charge on neutral and acidic polysaccharides can be modified by addition of base to ionize hydroxyls before chromatography. The basic conditions used control the level of N-acetylation, and the polysaccharides can be re-acetylated as needed. Borate salts can be used to enhance separation during ion-exchange chromatography. A combination of precipitation, filtration, and chromatographic procedures can also be used.
Diafiltrations, ultrafiltrations, and intermediate drying steps can be used as needed to concentrate polysaccharides while removing low molecular weight impurities or replacing processing salts and solvents. The precipitates or column fractions can be further purified using suitable methods (e.g., enzyme treatments, solvent extractions, or column chromatography) to remove impurities such as nucleic acids, proteins, and lipopolysaccharides. A preliminary side group modification can also be included in the purification process (e.g., de-O-acetylation, partial depyruvylation).
The final purification step can consist of buffer exchange and filtration followed by storage of purified liquid polysaccharide (frozen) or additional final precipitation and washing of the precipitate with solvent before drying followed by storage. Drying can be performed via several types of processes (e.g., drying under vacuum or in desiccators or by lyophilization). Drying of polysaccharides can be performed in desiccators (at various temperatures) and can include several steps of grinding or fluffing and return to the desiccators for further drying. Lyophilization of polysaccharides is possible with appropriate controls if the process requires retention of bound water. Some polysaccharides may require a residual amount of moisture to maintain stability over time. The polysaccharide is then stored under suitable conditions to avoid moisture uptake.
In-process Controls
Manufacturers identify critical process steps and perform appropriate tests to monitor the purification process. Among the latter are filter integrity tests, Microbiological Examination of Nonsterile Products: Microbial Enumeration Tests 61, Bacterial Endotoxins Test 85, and other suitable tests for residues of reagents (e.g., residual reagents, solvents, enzymes, or cations) used in purification. Polysaccharide size can be influenced throughout the production process from fermentation conditions to drying conditions or by the action of mechanical stirrers, impellers, or filtration devices. If molecular size is a critical quality attribute, analysts can perform an in-process test for size (e.g., high performance size-exclusion chromatography coupled with multi-angle laser light scattering [HPSEC-MALLS]) at the appropriate process steps to monitor and control polysaccharide size. In order to demonstrate process performance and reliability, manufacturers should characterize inherent residual contaminants (e.g., protein, DNA, and endotoxins). When validation studies have demonstrated removal of residual reagents, testing of purified polysaccharides can be omitted. If material must be sterile, analysts can perform Sterility Tests 71.
Protein Purification
The classes of bacterial protein vaccines include toxoids, nontoxoids (e.g., pertussis antigens), naturally occurring mutants (e.g., CRM197) as carrier proteins, and engineered recombinant products.
Toxoids. At the end of fermentation, toxin-containing culture medium should be separated aseptically from the bacterial mass as soon as possible or placed in a cold room until separation can be effected. The toxin content (Lf/mL) is checked by flocculation assay using the appropriate antitoxin standard to monitor production consistency (culture should contain NLT 40 Lf/mL). The toxin is purified first to remove any components that could cause adverse reactions in humans. A typical process includes depth filtration followed by 0.2-µm filtration to assist in removal of cellular debris. Following preliminary purification, the toxin is then detoxified with formaldehyde or glutaraldehyde or any suitable chemical reagent by a method that avoids both destruction of the immunogenic potency of the toxoid and reversion of the toxoid to toxin, particularly on exposure to heat. Some toxoids require a single addition of formaldehyde, but others can require multiple additions. Alternatively the toxin could be detoxified and then purified or partially purified by depth filtration, detoxified by addition of an appropriate aldehyde, filtered using 0.2-µm filtration, and then pooled. The pooled toxoid solution is further purified by clarification with activated carbon, followed by multiple ammonium sulfate precipitation steps that further fractionate and concentrate the toxoid. Typical additional purification steps include concentration, diafiltration, and/or chromatography. Purification before detoxification results in a purer product and can be advantageous if the toxoid is to be used as the protein component of a protein–carbohydrate conjugate (because copurifying high molecular weight glycans will be removed before detoxification).
At the end of fermentation, toxin-containing culture medium should be separated aseptically from the bacterial mass as soon as possible or placed in a cold room until separation can be effected. The toxin content (Lf/mL) is checked by flocculation assay using the appropriate antitoxin standard to monitor production consistency (culture should contain NLT 40 Lf/mL). The toxin is purified first to remove any components that could cause adverse reactions in humans. A typical process includes depth filtration followed by 0.2-µm filtration to assist in removal of cellular debris. Following preliminary purification, the toxin is then detoxified with formaldehyde or glutaraldehyde or any suitable chemical reagent by a method that avoids both destruction of the immunogenic potency of the toxoid and reversion of the toxoid to toxin, particularly on exposure to heat. Some toxoids require a single addition of formaldehyde, but others can require multiple additions. Alternatively the toxin could be detoxified and then purified or partially purified by depth filtration, detoxified by addition of an appropriate aldehyde, filtered using 0.2-µm filtration, and then pooled. The pooled toxoid solution is further purified by clarification with activated carbon, followed by multiple ammonium sulfate precipitation steps that further fractionate and concentrate the toxoid. Typical additional purification steps include concentration, diafiltration, and/or chromatography. Purification before detoxification results in a purer product and can be advantageous if the toxoid is to be used as the protein component of a protein–carbohydrate conjugate (because copurifying high molecular weight glycans will be removed before detoxification).
During detoxification and purification, endotoxin testing according to Bacterial Endotoxins Test 85, and formaldehyde, protein, and irreversibility testing are performed to control and ensure consistency of the purification process. If material must be sterile, Sterility Tests 71 can be performed.
Proteins/Recombinant Proteins.
Proteins used to make vaccines can be recombinant (in their native state or engineered to modify certain amino acids), or they can be naturally occurring mutants that have no wild-type activity yet are capable of inducing the appropriate immune response. Proteins are harvested from the fermenter and are extracted (e.g., by mechanical and chemical disruption) then purified by suitable methods, typically consisting of filtration–concentration steps (e.g., ultrafiltration, tangential-flow filtration, diafiltration, centrifugation, selective precipitation, and even direct capture using expanded-bed chromatography or big-bead technologies). The enriched protein solution can be further purified using appropriate filtration and chromatographic steps. For all equipment that contacts drug substances (e.g., chromatographic resins, membranes, disposable bag systems, or process lines), manufacturers should assess extractables and leachables. Analysts should determine column resin life for all chromatographic systems used in the purification (including number of uses, reconditioning requirements, and storage conditions).
The type of chromatography used to purify proteins depends on the physical/chemical properties of the desired protein as well as those of other molecular entities in the harvest culture. As an example, CRM197 can be purified using a multistep chromatographic process: Production material is first diafiltered and then is separated by ion-exchange chromatography (DEAE-Sepharose) in order to purify the target protein from other molecular entities present in the purification stream. The peak of interest is collected, and ammonium sulfate is added, followed by 0.22-µm filtration to condition the material before loading on the hydrophobic-interaction chromatography column (Phenyl Sepharose) for purification of the target protein based on its surface hydrophobicity. The peak fraction is then diluted with Water for Injection and is separated onto a ceramic hydroxyapatite column to further purify the target protein based on its surface charge. The eluted peak is then buffer exchanged into the storage buffer by ultrafiltration/diafiltration using cross-flow membrane filtration followed by 0.22-µm filtration to yield the sterile purified concentrate.
In-process control of protein purification includes monitoring specific protein content and critical process steps as well as monitoring removal of unwanted fermentation and purification components. The pH is critical for ion-exchange chromatography, and therefore pH should be monitored. For steps designed to remove endotoxin, procedures in Bacterial Endotoxins Test 85 are used to monitor column eluants. Bioburden is monitored according to Microbiological Examination of Nonsterile Products: Microbial Enumeration Tests 61 after filtration and chromatography steps. If material must be sterile, the sterility test in Sterility Tests 71 should be performed.
Polysaccharide–Protein Conjugates.
Polysaccharide preparation and activation polysaccharides used in conjugation reactions vary in size from native high molecular weight polysaccharide to oligosaccharides produced by controlled depolymerization. Some polysaccharides can also be modified (e.g., de-O-acetylated, partially depyruvylated). Sizing/depolymerization of polysaccharides is performed in a variety of ways (e.g., acid/base catalysis, chemical oxidation/reduction, microfluidization, or mechanical treatment). Activation of polysaccharides can be performed by several different methods depending on the lability of particular epitopes under differing depolymerization conditions or the type of conjugate desired (e.g., neoglycoconjugate or lattice-type conjugate, use of a linker molecule or direct conjugation, or reductive amination). Appropriately sized and/or activated polysaccharides are purified by suitable methods that typically consist of various combinations of concentration and filtration (ultrafiltration/diafiltration) and chromatographic methods (size-exclusion chromatography, HIC).
The in-process testing performed to monitor the depolymerization and activation process depends on the process used. Typical control tests are pH monitoring and temperature monitoring of the sizing and activation reactions. The size of the polysaccharide during depolymerization can be followed by an appropriate chromatographic procedure (e.g., HPLC-SEC RI or MALLS). Testing of depolymerized polysaccharide for select functional groups (e.g., O-acetyl, N-acetyl, or pyruvyl groups) may be required and can be determined by nuclear magnetic resonance (see Nuclear Magnetic Resonance 761). In-process testing of the activated polysaccharide depends on the activation process used. For example, if reductive amination is used to attach a linker to the depolymerized polysaccharide, the control testing would include measurement of reducing activity (e.g., available reducing sugars), polysaccharide content (e.g., for determining the loading ratio in conjugation), and total and free linker content (e.g., for determining the number of active sites for conjugation). Depending on the activation and conjugation process used (i.e., immediate conjugation after activation), consistency in degree of polysaccharide activation may also be demonstrated as part of process validation or reflected by characteristics of the final conjugate bulk. The concentration/filtration steps of the purification process are monitored for conductivity to ensure removal of salts.
Conjugation.
The conjugation chemistry used determines the type of conjugate made (i.e., neoglycoconjugate or lattice). The conjugate is obtained by the covalent binding of activated polysaccharides to the carrier protein. Conjugates are purified by suitable methods designed to remove residual reagents used for conjugation as well as to remove unreacted polysaccharide and protein. The removal of residual reagents is confirmed by suitable tests or by validation of the purification process. Suitable tests are carried out to determine residues of reagents used during inactivation and purification. When validation studies have demonstrated removal of residual reagents, the test on conjugate polysaccharides can be omitted.
Appropriate chromatographic procedures (HIC, SEC) and/or filtration (ultrafiltration/diafiltration, tangential-flow filtration) are used to remove the unreacted polysaccharides, protein, residual chemicals, and salts that are used in conjugation or that are by-products of conjugation. Bioburden testing (see Microbiological Examination of Nonsterile Products: Microbial Enumeration Tests 61) is performed before sterile filtration.
INTERMEDIATES
An intermediate or process intermediate in vaccine manufacture is the reaction product of each step in the process except the last one, which forms the final product. Examples of intermediates are bulk-purified polysaccharides, proteins, and activated polysaccharides that conjugate to protein.
Most vaccine production processes are stepwise and take more than one elementary step to complete. An intermediate is produced from raw materials at one or more process steps (e.g., bacterial growth, extraction and purification, and chemical modification), eventually resulting in the drug substance. The identification of the key intermediates, their production, and sampling for analytical tests must be defined in controlled documents (e.g., batch records, analytical protocols).
Intermediates can be stored for considerable periods of time before further processing and can be included in a formal stability program (see Storage Stability, above). Stability studies in normal or accelerated conditions should be performed to define maximal hold time for intermediates and when significant process changes are implemented.
From raw material to finished drug substance, testing throughout the process ensures a quality product. Testing of intermediates is a key quality control step to ensure their identity and purity. The quality attributes of the intermediate are commonly tested in conjunction with further processing, and their release testing should be considered. Standard operating procedures (SOPs) must be properly defined for the analytical control tests. Because of their critical role in the production process, some key intermediates could be included in formal release testing, in addition to the intermediates identified for in-process testing.
Examples of tests for structural characterization of carbohydrate-based intermediates include the following:
- identity and O-acetylation level (nuclear magnetic resonance)
- molecular size and polydispersity (SEC–UV, SEC-Refractive Index, SEC-MALLS, SEC-Fluorescence)
- saccharide content [colorimetric assays, high-pH anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD), -Fluorescence]
-
specific rotation, see Optical Rotation 781
- saccharide content, O-acetyl content (colorimetric assays)
- counterion content, e.g., Na+ and Ca2+ (inductively coupled plasma–mass spectroscopy, atomic absorption).
Examples of tests for the purity of carbohydrate-based intermediates based on estimates of the product- and process-related impurities include the following:
-
endotoxin content (Bacterial Endotoxins Test 85)
- proteins, nucleic acids, proteins, cetavlon (colorimetric assays)
-
water content (Karl Fischer titration; see Water Determination 921)
- volatile substances (thermogravimetry)
-
organic solvents, e.g. ethanol, phenol, acetone, DMSO, or ethyl acetate (gas chromatography–flame ionization detection; see Residual Solvents 467)
-
bioburden (total viable aerobic count of microbial contamination; see Microbiological Examination of Nonsterile Products: Microbial Enumeration Tests 61)
Examples of tests for structural characterization and purity estimation of protein-based intermediates include the following:
- identity and molecular size (SDS-PAGE, Western Blot, SEC-UV, SEC-Fluorescence, SEC-MALLS, reverse phase chromatography)
-
endotoxin content (see Bacterial Endotoxins Test 85)
-
residual nucleic acids [colorimetric assay; see Nucleic Acid-Based Techniques—Approaches for Detecting Trace Nucleic Acids (Residual DNA Testing) 1130]
-
bioburden (total viable aerobic count of microbial contamination; see Microbiological Examination of Nonsterile Products: Microbial Enumeration Tests 61)
- other proteins as impurities (SDS-Page, Western Blot, SEC-UV, SEC-Fluorescence, reversed-phase chromatography)
The tests previously reported for protein-based intermediates are also applicable when the protein-based product is defined as the drug substance. In addition to the examples reported above, many other methodologies can be applied for the identity and purity evaluation.
Stability tests for intermediates can include physicochemical methods (see section on Intermediates, above), formally included within an analytical panel for the stability study. In addition, biological and immunochemical tests [e.g., enzyme-linked immunosorbent assay (ELISA)] can be included. Bioburden and endotoxin testing may not be required at each level (each intermediate, drug substance) provided testing is performed at sufficient steps in the overall production process. Bioburden is typically performed prior to sterile filtration via in-process testing. If intermediates must be stored and/or subsequently shipped to a different location for further processing, the stability of these materials must be demonstrated.
DRUG SUBSTANCE
The drug substance is the final bulk that contains the antigen at the desired concentration and is ready for the addition of other ingredients (e.g., diluents, bulking agents, stabilizing excipients, adjuvants, or preservatives) to produce the finished dosage formulation.
The drug substance is the final product of the antigen manufacture process, before the formulation of the final vaccine dosage. The final bulk may be prepared aseptically or may include a sterilization step. Sampling for analytical tests for release and stability studies (see Storage Stability, above) must be defined in controlled documents (e.g., batch records, analytical protocols).
Drug substances can be stored for a considerable period of time before further processing, but if it is stored the drug substance must be included in a formal stability program (see Storage Stability, above). Stability studies in normal or accelerated conditions should be performed to define maximal hold times. A stability program is required for formal stability studies, and the studies must be executed according to a protocol that contains detailed information about types of tests, specifications, testing intervals, and time points.
Testing of the drug substance must be performed to ensure its identity and purity. All the testing must be done according to established SOPs, and all tests must have specifications (or provisional specifications, where applicable).
Examples of tests for structural characterization of carbohydrate-based products include the following:
- identity and O-acetylation level [nuclear magnetic resonance (NMR)]
- total and free saccharide content [HPAEC-PAD, capillary electrophoresis (CE), or colorimetric assays]
- total and free protein content (colorimetric assay, SEC with UV, RI, or fluorescence detection, CE)
- O-acetyl content (colorimetric assays)
- molecular size (SEC-UV, -RI, -Fluorescence, or -MALLS)
Examples of tests for the purity of carbohydrate-based drug substances based on estimating the product- and process-related impurities include the following:
-
endotoxin content (Bacterial Endotoxins Test 85)
- process-related residuals not shown to be removed by process validation (e.g., cyanide, iodate, bromide, ammonium sulfate, or organic solvents)
-
Bioburden (total viable aerobic count of microbial contamination; see Microbiological Examination of Nonsterile Products: Microbial Enumeration Tests 61)
In addition to the examples reported above, many other methodologies can be applied for identity and purity evaluation. For instance, specific impurities that must be measured are determined by negotiations between manufacturers and the national drug regulatory agency during the licensure process. Bioburden and endotoxin testing may not be required at each level (each intermediate, drug substance) provided testing is performed at sufficient steps in the overall production process. Bioburden is typically performed prior to sterile filtration via in-process testing.
All the results must be reported in a controlled document. Stability tests can include both physicochemical methods (see stability information, above) and biological/immunochemical tests (e.g. ELISA and SBA; see Quality of Biotechnological Products: Stability Testing of Biotechnological/Biological Products 1049).
DRUG PRODUCT AND LOT RELEASE
General principles are described in Vaccines for Human Use—General Considerations 1235, which outlines the lot release procedure in accordance with 21 CFR 610.1 and 21 CFR 610.2. For products that will be used in the United States, samples and protocols containing all appropriate tests must be submitted to FDA for review and/or testing. If FDA determines that the lot meets the standards of safety, purity, and potency required for the particular vaccine, the lot is approved for release, distribution, and marketing.
Tests required for each lot-release protocol include potency, general safety, sterility, purity, identity, and constituent materials. Potency and potency-related tests are different for each bacterial vaccine. The inclusion of these tests makes each bacterial vaccine lot-release protocol unique.
The contents of a final container of each filling of each lot are tested for identity after labeling is completed. Identity is established either by physical or chemical characteristics of the vaccine, inspection by macroscopic or microscopic methods, specific cultural tests, or in vivo or in vitro immunological tests. In large part, identity testing is performed to distinguish the vaccine from other materials that are manufactured at the same site (21 CFR 610.14). The same tests that establish identity may also be appropriate for defining the quantity of immunogen present in the final vial. This is especially important for carbohydrate-based vaccines that are dosed by mass and for which physicochemical measures of antigen quality are used.
Immunochemical methods, which include immunoprecipitation methods and immunoelectrophoretic methods, have been useful. Immunoprecipitation methods, flocculation and precipitation, can be carried out in solution or in a gel matrix and involve mixing the antigen with an appropriate antibody, leading to the formation of flocculating or precipitating aggregates that can be detected visually or by light scattering (light scattering or nephelometry). The ratio of reactants must be varied to optimize the detected response. In solution this can be achieved by titrating one reactant with the other, and increased sensitivity can be obtained by using antigen- or antibody-coated particles (e.g., latex) as reactants. In gel systems, a gradient is created as one or more of the reactants diffuse, creating a visible line where precipitation occurs. Immunoelectrophoresis (IE) is a qualitative technique that combines two methods: gel electrophoresis followed by immunodiffusion. Crossed IE is a modification of the IE method that is suitable both for qualitative and quantitative analysis. Visualization and characterization of immunoprecipitation lines can be performed by selective or nonselective stains, fluorescence, enzyme or isotope labeling, or other relevant techniques. Selective staining methods are usually performed for characterization of nonprotein substances in the precipitates. In translucent gels, such as agar or agarose, the precipitation line becomes clearly visible in the gel provided that the concentration of each of the reactants is appropriate.
Where multiple active components are present as a result of copurification (e.g., certain acellular pertussis vaccines), the manufacturer must demonstrate that the composition of the product is consistent between batches, unless this has been validated during the development of the manufacturing process.
For certain vaccines, notably those that use purified polysaccharide immunogens, identity and immunogen quantity can be demonstrated using one or more chemical and physicochemical approaches such as colorimetric determinations of different groups of sugar residues expected to be present, chromatography, or high-field nuclear magnetic resonance spectroscopy.
A number of classical colorimetric assays for quantification of various classes of sugar have been used to define the composition of polysaccharides used as vaccines, including the orcinol assay for ribose, phosphorus, sialic acid, uronic acids, and aminosugars. In general, these approaches have been superseded by chromatographic methods, including gas chromatography and HPAEC, which is widely used to determine Hib PRP glycoconjugate immunogens in monovalent or combination vaccines and in meningococcal conjugate immunogens.
Immunochemical methods that have been used to quantify polysaccharide antigens include (a) rocket immuno-electrophoresis and (b) rate nephelometry. Electroimmunoassay, also called rocket immuno-electrophoresis, is a quantitative method to determine antigens that differ in charge from the antibodies. Electrophoresis of the antigen to be determined is carried out in a gel that contains a lower concentration of the corresponding antibody. Nephelometry methods have been used to quantify antigen in pneumococcal conjugate vaccines.
The consistency of the molecular size and molecular size distribution of polysaccharide- and carbohydrate-containing conjugate vaccines can be determined by gel-permeation chromatography on appropriate resins calibrated with suitable molecular weight markers or coupled to laser light-scattering equipment that indicates absolute molecular weight if a value for the refractive index increment (dn/dc) is known. Measurement of the molecular size of formulated conjugates may not be feasible for multivalent glycoconjugate vaccines. Integrity of the conjugate may be demonstrated by alternative, product-specific methods. Another alternative to demonstrate integrity of the glycoconjugates in the final product is measurement of molecular size as part of the stability studies at monovalent conjugate bulk prior to formulation of the multivalent vaccine.
Unless the contrary has been validated, manufacturers should demonstrate that reversion to toxicity has not occurred (and will not occur over the shelf life) for a product derived from or containing a toxoid material. This may require the use of a cell line or an in vivo test, although enzymatic approaches are being validated.
An antigenic purity test is an assay that assesses the quantity of antigen and is used for diphtheria and tetanus toxoid vaccines. The antigen content is determined by a flocculation assay.
The manufacturer should prove a high and consistent level of immunogen adsorption to any solid-phase adjuvant (such as aluminum phosphate or aluminum hydroxide) that is consistent with the release specification.
For certain vaccines such as the anthrax vaccine and toxoid vaccines, the manufacturer is required to demonstrate that the vaccine is protective against disease or death in animal models challenged with a predefined dose of the target pathogen. This generally requires definition of the animal model, route of administration, vaccine dilutions required, a means to observe effects, and a reference vaccine against which effects are compared. The data should be analyzed appropriately (see Analysis of Biological Assays 1034).
Stability-indicating assays are those used to determine the stability of the product. Of primary importance is the potency assay, although glycan degradation may be important in glycoconjugate vaccines.
Other Vaccine Components and Vaccine Properties.
Aluminum compounds are the primary adjuvants used in vaccines in the United States. General chapter Vaccines for Human Use—General Considerations 1235 provides provisions of the 21 CFR 610.15 governing the use of aluminum and amounts allowed. The adjuvants widely used in bacterial vaccines include aluminum potassium sulfate (alum), aluminum phosphate, aluminum hydroxide, and combinations of these compounds. Bacterial vaccines formulated with such adjuvants are referred to as adsorbed vaccines, and this term may be included in the official name of the vaccine. Other adjuvant systems may be evaluated. Aluminum is quantitated using colorimetric, titrimetric, emission or atomic absorption spectroscopy, or inductively coupled plasma–mass spectrometry.
For regulations regarding residual manufacturing reagents, see the FDA's 1999 Guidance for Industry: Content and Format of Chemistry, Manufacturing, and Controls Information and Establishment Description Information for a Vaccine or Related Product. Manufacturing reagents such as formaldehyde and glutaraldehyde sometimes are used in inactivation, the toxoid-making processes, or elsewhere during manufacture and may be present in residual amounts in the final product. Limits of formaldehyde and other residuals must be minimized in accordance with the approved product license.
Common preservatives used in bacterial vaccines include thimerosal, phenol, 2-phenoxyethanol, and benzalkonium chloride. Vaccines for Human Use—General Considerations 1235 and 21 CFR 610.15 provide additional information about the minimization of thimerosal content and the production of thimerosal-free vaccines. Limits and content specifications are set for each bacterial vaccine in the product license.
Each lot of final containers of a vaccine intended for use by injection is tested for bacterial endotoxins as indicated in Bacterial Endotoxins Test 85.
Each lot of final containers of a vaccine intended for use by injection may be tested for pyrogenic substances as indicated in Pyrogen Test 151 and 21 CFR 610.14.
Each lot of dried product shall be tested for residual moisture (see Loss on Drying 731 and FDA Guideline for the Determination of Residual Moisture in Dried Biological Products. January 1990). Residual moisture should be determined for lyophilized vaccines.
A general safety test is performed on biological products intended for administration to humans with the purpose of detecting extraneous toxic contaminants. Procedures and exceptions are specified in 21 CFR 610.11.
Excipient identity and quantity, preservatives, diluents, adjuvants, extraneous protein; and cell culture-produced vaccines and antibiotics are tested according to 21 CFR 610.15 and/or appropriate guidance documents.
“Free” or unconjugated saccharide in glycoconjugate vaccines is considered undesirable and is subject to limit specifications. As an alternative to controlling free or unconjugated saccharide, integrity of the conjugates in the final product may be demonstrated via an appropriate method, said method depending on the properties of the final product (composition, adsorption, etc). A test method that measures the increase in the amount of free saccharide is a stability-indicating method. The methods adopted depend on separation of saccharide from conjugate and application of the methods above to quantify the unconjugated saccharide. Separation methods used include membrane separation (such as dialysis), use of hydrophobic media to specifically trap the conjugate, solvent extraction, and selective immunochemical precipitation of the conjugate using anticarrier antibodies.
The sterility of each lot of each product is conducted according to procedures described in Sterility Tests 71 and 21 CFR 610.12 for both bulk and final container material.
Information insert (Label). Vaccine product labeling is regulated in compliance with 21 CFR 201 and 610. Requirements are set both for container labeling and package labeling.
OTHER REQUIREMENTS
Retention samples are held by the manufacturer for at least six months after the expiration date. Enough material of each lot of each product is held for examination and testing for safety and potency (see 21 CFR 600.13).
Records are maintained concurrently with each step in the manufacture and distribution of product such that at any time successive steps of manufacture and distribution may be traced (see 21 CFR 600.12).
For storage conditions, see 21 CFR 610.50 and 53.
For shelf life/expiry date, see 21 CFR 610.50 and 53.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Tina S. Morris, Ph.D.
Vice President, Biologics & Biotechnology (301) 816-8397 |
(GCBA2010) General Chapters - Biological Analysis |
USP38–NF33 Page 1570
Pharmacopeial Forum: Volume No. 37(4)