



INTRODUCTION
Nuclear magnetic resonance (NMR) spectroscopy is an analytical method based on the magnetic properties of certain atomic nuclei. As is the case with other types of spectroscopy, absorption or emission of electromagnetic energy at characteristic frequencies provides structural information. NMR differs from other types of spectroscopy because the discrete energy levels between which the transitions take place are present only when the nuclei are placed in a magnetic field.
Although widely recognized as one of the most powerful structure-elucidation tools available, with proper experimental design, it can also be used for accurate qualitative and quantitative measurements. See general information chapter Applications of Nuclear Magnetic Resonance Spectroscopy  1761
1761 . [Note—Above 1000 chapters are for informational purposes only. ]
. [Note—Above 1000 chapters are for informational purposes only. ]
QUALIFICATION OF NMR INSTRUMENTS
Qualification of an NMR instrument can be divided into three elements: Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ). For further discussion, see general information chapter Analytical Instrument Qualification 1058.
Installation Qualification
The IQ requirements provide evidence that the hardware and software are installed to accommodate safe and effective use of the instrument at the desired location.
Operational Qualification
In OQ, an instrument's performance is characterized using standards to verify that the system operates within target specifications. The purpose of OQ is to demonstrate that instrument performance is suitable for a given application. Because so many different approaches are available for measuring NMR spectra, OQ using standards with known spectral properties is recommended. Generally, sealed NMR tubes are available as reference standards for measuring signal-to-noise (S/N) and lineshape.
Performance Qualification
PQ helps to determine that the instrument is capable of meeting the user's requirements for all critical-to-quality (CTQ) measures. PQ documentation should describe the following:
- The definition of the specific performance criteria and detailed test procedures including test samples and instrument parameters.
- The elements that will be measured to evaluate the criteria and the predefined specifications.
- The test interval, which may be time-of-use.
- The use of bracketing samples or groups of samples.
- The defined corrective actions that will be implemented if the spectrometer does not pass the specifications.
Periodic PQ should include a subset of the OQ tests to ensure that those aspects of the instrument that are being supplied are performing at a level that produces data that are suitable for its intended use. Depending on typical use, the specifications for PQ may be higher or lower than the manufacturer's installation specifications. Typical CTQs include S/N ratio and resolution tests for all nuclei of interest. Method-specific PQ tests, also known as system suitability tests, may be used in lieu of PQ requirements for validated procedures.
The PQ samples and tests in the following subsections are typical examples only. Other tests and samples may be used to establish specifications for specific purposes. Instrument vendors often provide samples and test parameters that can be used as part of the PQ package.
resolution and lineshape measurement—1h nmr (see Figure 1)
Sample:
1% chloroform in acetone-d6 ( 500 MHz), 3% chloroform in acetone-d6, degassed and sealed
500 MHz), 3% chloroform in acetone-d6, degassed and sealed
Spectral width:
< 1 KHz
Data acquisition time:
NLT 10 s
Tip angle:
90
Relaxation time:
60 s
Spinning rate:
Static or 20 Hz
Pulse sequence:
Delay-pulse-acquire with no decoupling
Processing:
No line broadening, zero-filling to 128 k
+'&img=../../images/v38332/c761-f1-pf362.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 1. 1H NMR spectrum of chloroform in acetone-d6 obtained at 400 MHz. The linewidth measured at 0.55% and 0.11% of the 13C satellites was 2.7 and 5.5 Hz, respectively.
Shim the magnet with special attention to the off-axis shims, acquire a single acquisition, phase to pure absorption, and measure the linewidth at 50%, 0.55%, and 0.11% maximum intensity. The linewidth should pass specifications at these positions, and, in addition, the lineshape should be Lorentzian. On modern NMR spectrometers, the lineshape is frequently obtained on a nonspinning sample because the off-axis shims can be set so well that there is essentially no difference between spectra obtained spinning and nonspinning. In addition, two-dimensional spectra should be obtained on a static sample.
s/n measurements—1h nmr (see Figure 2)
Sample:
0.1% ethylbenzene in chloroform-d, 1% ethylbenzene in chloroform-d (< 200 MHz) degassed and sealed
Spectral width:
10 ppm
Data acquisition time:
400 ms
Tip angle:
90
Relaxation delay:
60 s
Spinning rate:
0 or approximately 20 Hz
Pulse sequence:
Delay-pulse-acquire with no decoupling
Processing:
Exponential with 1-Hz line broadening
Referencing:
Tetramethylsilane (TMS) = 0.0 ppm or the center of the quartet = 2.65 ppm
+'&img=../../images/v38332/c761-f2-usp36.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 2. 1H NMR spectrum of 0.1% ethylbenzene obtained at 400 MHz with an S/N ratio of 550:1
The concentration of ethylbenzene should be chosen to achieve S/N ratio specifications in the range of 20–1000. Concentrations that typically result in measurements outside that range are of limited utility in assessing the performance of the instrument. Nevertheless, established standard solutions are conventionally used. The magnet should be shimmed as well as possible. Ideally, this test should be run immediately after the lineshape test because most of the shims will be nearly maximized. Acquire a single acquisition, phase the spectrum in pure absorption mode, and measure the S/N of the ethylbenzene quartet. This experiment can be run with or without sample spinning. With a spinning sample, the S/N value that is measured should be only about 10% higher than that obtained with a nonspinning sample if the off-axis shims are well adjusted. A higher ratio would indicate that the determination would benefit from further shimming with the off-axis shims.
Most modern spectrometers have software that perform the S/N measurement after the operator has identified the signal and noise regions. Manual calculations can also be made. Measure the amplitude (A) from the center of the baseline to the peak of the highest of the central two lines in the quartet. Measure the peak-to-peak noise height (H) from the lowest noise peak to the highest noise peak in the 3–5 ppm region. The noise may be vertically multiplied by a factor for accurate measurement of high S/N spectra. Calculate the S/N as follows:
S/N = k × 2.5 × A/H [1]
[1]
where k is the vertical expansion factor of the noise region used. The factor of 2.5 converts the peak-to-peak S/N to root-mean-squared (rms) noise, which is the standard convention for reporting S/N in NMR spectroscopy. Computerized S/N calculations can be used provided the specifications are set and tested by the same procedure. At the discretion of the spectroscopist, an S/N value lower than that specified by the manufacturer may be used if it is judged to be sufficient for the current application.
s/n measurements 13c nmr (see Figure 3)
Sample:
40% p-dioxane in benzene-d6 (v/v) (degassed and sealed)
Spectral width:
Approximately 200 ppm
Tip angle:
90
Relaxation delay:
300 s
Spinning rate:
Approximately 20 Hz
Pulse sequence:
Delay-pulse-acquire with no decoupling
Processing:
Exponential with 3.5-Hz line broadening, zero-filling to 32k
Referencing:
TMS = 0.0 ppm or the center of the benzene triplet = 128.4 ppm
+'&img=../../images/v38332/c761-f3-pf362.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 3. 13C NMR spectrum of the ASTM standard 40% p-dioxane in benzene-d6 (v/v) obtained at 100.6 MHz, with an S/N ratio of 140:1
With a well-shimmed magnet, acquire a single acquisition following a minimum delay of 300 s, phase the spectrum in pure absorption mode, and measure the height of the benzene triplet at approximately 128.4 ppm from the center of the baseline. The peak-to-peak noise can be measured as above with appropriate vertical expansion of 80–120 ppm. S/N calculations can be made as in Equation 1 or by computer calculation.
The benzene-d6 triplet has no nuclear Overhauser enhancement (NOE). Consequently, this test verifies the performance of only the 13C channel.
performance of both of the 13c and 1h channels (see Figure 4)
Sample:
up to 10% ethylbenzene in chloroform-d (degassed and sealed)
Spectral width:
200 ppm
Data acquisition length:
64k points
Tip angle:
90
Relaxation delay:
300 s
Spinning rate:
Approximately 20 Hz
Pulse sequence:
Delay-pulse-acquire with composite pulse decoupling set to the center of the 1H spectrum
Processing:
Exponential with 0.3-Hz line-broadening
Referencing:
TMS = 0.0 ppm or the center of the chloroform-d, triplet = 77.23 ppm
+'&img=../../images/v38332/c761-f4-usp36.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 4. 13C NMR spectrum of 10% ethylbenzene obtained using a cryogenically cooled dual 1H/13C probe at 150.9 MHz, with an S/N ratio of 640:1
The shimming should be sufficient to pass the resolution and lineshape tests described above. The measurement of S/N is done from the peak height of the larger resonance of the two near 128 ppm. The noise is measured as above in the region of 80–120 ppm, with appropriate vertical expansion. S/N is calculated by the computer or as in Equation 1.
relaxometry measurements—low field-nmr (lf-nmr)
The PQ should be performed before the collection of experimental data.
Dissolve an accurately weighed quantity of manganese (II) chloride tetrahydrate (MW 197.91) in water, and quantitatively dilute with water to obtain check solutions that have known concentrations of 0.9, 2.7, and 4.5 mM.
Place a portion of each of the solutions into sample holders suitable for the configuration of the specific model of the LF-NMR spectrometer. Warm to 40 for NLT 10 min, and measure the spin-lattice relaxation time (T1) of water. The average T1 for replicate measurements must be within 5% of 156, 52, and 32 ms for the 0.9, 2.7, and 4.5 mM solutions, respectively.
Characterizing Instrument Performance
Specific procedures, acceptance criteria, and time intervals for characterizing NMR spectrometer performance depend on the instrument and its intended application. Many NMR applications use previously validated experiments that relate NMR spectra to a physical or chemical property of interest. Stable instrument performance over extended periods of time should be demonstrated. This practice provides some assurance that reliable measurements can be taken from sample spectra using previously validated NMR experiments.
QUALITATIVE AND QUANTITATIVE NMR ANALYSIS
NMR spectroscopy has been used for a wide range of applications such as structure elucidation; thermodynamic, kinetic, and mechanistic studies; and quantitative analysis. Some of these applications are beyond the scope of compendial methods.
All characteristics of the signal—chemical shift, multiplicity, linewidth, coupling constants, relative intensity, and relaxation time—contribute analytical information.
Qualitative Applications
Comparison of a spectrum from the literature or from an authentic standard with that of a test sample may be used to confirm the identity of a compound and to detect the presence of impurities that generate extraneous signals. The NMR spectra of simple structures can be adequately described by the value of the chemical shifts and coupling constants, and by the relative number of nuclei represented by the integral of each signal. (The software of modern instruments have available programs that generate simulated spectra using these data.) Experimental details, such as the solvent used, and the chemical shift reference, must also be provided.
For unknown samples, NMR analysis, usually coupled with other analytical techniques, is a powerful tool for structure elucidation. Chemical shifts provide information on the chemical environment of the nuclei. Extensive literature is available with correlation charts and rules for predicting chemical shifts. The multiplicity of the signals provides important structural information. The magnitude of the scalar coupling constant, J, between residual protons on substituted aromatic, olefinic, or cycloalkyl structures is used to identify the relative position of the substituents. Routine 13C spectra are obtained under proton decoupling conditions that remove all heteronuclear 13C-1H couplings. As a result of this decoupling, the carbon signals appear as singlets, unless other nuclei that are not decoupled are present (e.g., 19F, 31P).
Chemical exchange is an example of the effect of intermolecular and intramolecular rate processes on NMR spectra. If a proton can experience different environments by virtue of such a process (tautomerism, rotation about a bond, exchange equilibria, ring inversion, etc.), the appearance of the spectrum will be a function of the rate of the process. Slow processes (on an NMR time scale) result in more than one signal from the interconverting species; fast processes average these signals to one line; and intermediate processes produce broad signals, which sometimes cannot be easily found in the spectra.
The software of modern FT-NMR spectrometers allows for sequences of pulses much more complex than the repetitive accumulation of transients described above. Such experiments include homonuclear or heteronuclear multidimensional analysis, which determines the correlation of couplings and may simplify the interpretation of otherwise complex spectra.
See chapter 1761 for detailed descriptions of common two-dimensional experiments.
Quantitative Applications
I. General considerations of quantitative NMR: see
II. The scope of this section is limited to quantitation by one-dimensional NMR. Although any of the NMR active nuclei can be used to obtain quantitative data, the discussion here will be limited to 1H. There are two kinds of quantitation by NMR: relative and absolute.
- Relative quantitation involves measurement of relative amounts of species in a sample based on integration of peaks due to each of the components measured. The integrals are normalized by factor N, that is, the integral is divided by the number of equivalent nuclei represented by that peak to give the relative molar concentration of each component.
-
Absolute quantitation is the direct measurement of the actual amount of analyte independent of other components contained in that sample. There are two basic methods for absolute quantitation based on the kind of reference standard that is used to calibrate the NMR signal.
-
Internal reference standard
- Definition: The reference standard is co-dissolved in the analyte test solution.
-
Procedure
Typical NMR solution preparation: An NMR solution is prepared with exact weights of both the analyte and reference standard. The largest source of error in this quantitative NMR method is from weighing, so the use of larger weights is recommended to minimize this error. This quantitative method is based on a comparison of the reference standard and analyte NMR peaks and their respective concentrations. Since the analyte and reference standard are in the same solution, the analyte and reference standard are contained in the same volume, and only their masses are compared. Therefore, the exact volume is not required. Typically, at least three replicates are prepared.
Data acquisition: Data is acquired under quantitative conditions, see1761. For example, the pulse repetition time should be at least 5 times the longest T1 when a 90 pulse is used.
Data processing: Process the data, using zero-filling if necessary, such that a sufficient number of points define a peak. For example, experience has shown that at least 16 points gives a good quantitative representation of a peak.
Analysis: Integrate appropriate peaks. For example, avoid using peaks that are overlapped, or due to hydrogens capable of exchanging. The determination of the amount of analyte derives from the basic proportionality between the peak intensity and the concentration of the solute.where I = integral; N = normalization factor; and [ ]1H = 1H relative molar concentration, and the subscripts A and RS represent the analyte and reference standard, respectively.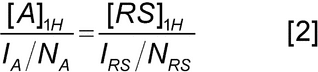 The mass of the analyte is thus calculated according to the following equation.
The mass of the analyte is thus calculated according to the following equation.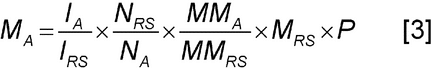 where MA = mass of the analyte, MM = molar mass, and P = purity of the reference standard.
where MA = mass of the analyte, MM = molar mass, and P = purity of the reference standard. -
A common application of absolute quantitation is the determination of the purity of a sample. The weight % purity is given by
where MS is the total mass of the sample with contributions from the analyte plus any contaminants that may be present in the sample such as water and salts. Combining Equations 3 and 4, the weight % purity is given by
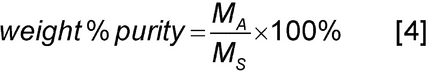
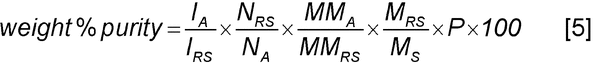
-
External reference standard
- Definition: The classical external reference standard method consists of solutions of a reference standard and analyte that are each in separate NMR tubes. One variation of an external reference standard is the standard test solution contained in a coaxial tube and is inserted into an analyte test solution contained in an NMR tube. Another variation is the introduction of a computer-generated signal into the spectrum of a reference standard solution of known concentration to calibrate the signal's response (intensity per 1H molar concentration, in the case of 1H NMR), followed by insertion of that calibrated computer-generated signal into the spectrum of an analyte test solution. This section will address the use of an external reference in the classical sense.
-
Procedure
NMR solution preparation: NMR solutions of known concentrations of each of the analyte and reference standard are prepared using exact weights and volumes. Again, the use of larger weights is recommended to minimize the weighing error. Typically replicates of the analyte solutions and reference standard solutions are prepared. The analyte and reference must be prepared in the same solvent to minimize probe tuning differences.
Data acquisition and processing: Same as in II.B.1.b, internal reference standard. Apply the same acquisition and processing parameters to the analyte and reference standard spectra.
Analysis: Integrate appropriate peaks in the spectra of the analyte and reference standard. The amount of analyte is calculated according to the following equation:where V = volume.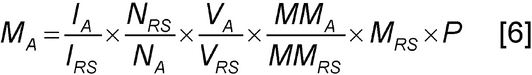 Application to weight % purity: Weight % purity values may be similarly calculated as in Equation 5.
Application to weight % purity: Weight % purity values may be similarly calculated as in Equation 5.
-
Internal reference standard
-
The internal and external reference standard methods each have their own set of advantages and disadvantages.
- Chemical interactions: Preparation of the reference standard and the test material in separate solutions avoids chemical interactions between the test sample and reference standard that may otherwise occur with an internal reference standard.
- Spectral overlap: The use of an external reference standard also avoids potential overlap between peaks of the reference standard and test sample that can occur with an internal standard.
- Calibration: Once an NMR response has been calibrated with external reference standard solutions, this calibration may be applied to any other sample in the same solvent given that i) the instrument has been demonstrated to be stable over the time between when the calibration is done and when data is acquired on the test material, ii) system suitability has been established on the day that the measurement on the test material is made, and iii) absolute integrals are compared. In the case of internal reference standards, the measurement on the reference standard and test sample is made under absolutely identical conditions.
- Accuracy and precision: Multiple external reference standard solutions may be prepared to average the errors in the mass and volume measurements during sample preparation, thereby improving the accuracy of the calibrated NMR response. In the case of internal reference standards, single measurements of the reference standard and analyte are made for each replicate test solution. The combined errors from the mass measurements of the reference standard and test sample as well as instrumental electronic variations determine the standard deviation of the average MA or weight % purity values.
+'&img=../../images/v38332/c761_eq2_pf376.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
+'&img=../../images/v38332/c761_eq3_pf376.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
+'&img=../../images/v38332/c761_eq4_pf376.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
+'&img=../../images/v38332/c761_eq5_usp36.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
+'&img=../../images/v38332/c761_eq6_pf376.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
VALIDATION AND VERIFICATION OF NMR ANALYTICAL PROCEDURES
If an NMR procedure is provided in a monograph, verification of suitability (see 1226) under actual conditions of use is required. Validation is required only when an NMR method is an alternative to the official procedure for testing an official article. The objective of validation of a procedure relying on the NMR method is to demonstrate that the measurement is suitable for its intended purpose, including the following: quantitative determination of the main component in a drug substance or a drug product (Category I assays), quantitative determination of impurities (Category II), and identification tests (Category IV). [Note—For a definition of the different categories, see Validation of Compendial Procedures 1225. ] Depending on the category of the test, analytical procedure validation requires the testing of specificity, linearity, range, accuracy, precision, quantitation limit, and robustness. These analytical performance characteristics apply to externally standardized methods and to the method of standard additions.
Chapter 1225 provides definitions and general guidance on analytical procedures validation without indicating specific validation criteria for each characteristic. The intention of the following sections is to provide the user with specific validation criteria that represent the minimum expectations for this technology. For each particular application, tighter criteria may be needed in order to demonstrate suitability for the intended use.
Analytical Procedure Validation
The objective of an analytical procedure validation is to demonstrate that the analytical procedure is suitable for its intended purpose by conducting experiments and obtaining results that meet predefined acceptance criteria. NMR analytical procedures can include the following: quantitative tests for major component and impurities content, limit tests for the presence of impurities, quantification of component in a product or formulation, and/or identification tests.
Performance characteristics that demonstrate the suitability of an analytical procedure are similar to those required for any analytical procedure. A discussion of the applicable general principles is found in chapter 1225. Specific acceptance criteria for each validation parameter must be consistent with the intended use of the analytical procedure.
The performance characteristics that are required as part of a validation for each of the analytical procedure categories is given below.
specificity
The purpose of a specificity test is to demonstrate that measurements of the intended analyte signals are free of interference from components and impurities in the test material. Specificity may be applied to all categories and is a requirement for Category IV. Specificity tests can be conducted to compare NMR spectra of other components and impurities that are known from synthetic processes and formulations and test preparations. For an identification NMR analytical procedure (Category IV), validation experiments may include multidimensional NMR experiments to validate correct assignments of chemical shifts and to confirm the structure of the analyte.
Validation criteria:
Specificity is ensured by use of a reference standard wherever possible and demonstrable lack of interference from other components.
linearity
A linear relationship is exhibited between the analyte concentration and instrument response; this should be demonstrated by measuring responses of analyte from NLT five standard solutions at concentrations encompassing the anticipated concentration range of analyte(s) of the test solution. For Category I, standard solutions can be prepared from reference materials in an appropriate NMR solvent. For Category II, NMR analytical procedures that are used to quantitate impurities, linearity samples can be prepared by spiking suitable test samples that contain low amounts of analyte or by spiking matrix samples at concentrations of the expected range. The standard curve should then be constructed using appropriate statistical analytical procedures such as a least squares regression. The correlation coefficient (R), y-intercept, and slope of the regression line should be determined. Absolute values determined for these factors should be appropriate for the procedure being validated.
Validation criteria:
The correlation coefficient (R) must be NLT 0.995 for Category I assays and NLT 0.99 for Category II quantitative tests.
range
The range between the low and high concentrations of analyte is given by the quantitative NMR analytical procedure. This is normally based on test article specifications in the USP monograph. It is the range within which the analytical procedure can demonstrate an acceptable degree of linearity, accuracy, and precision, and may be obtained from an evaluation of that analytical procedure. Recommended ranges for various NMR analytical procedures are given below.
Validation criteria:
For Category I tests, the validation range for 100.0% centered acceptance criteria is 80.0%–120.0%. For noncentered acceptance criteria, the validation range is 10.0% below the lower limit to 10.0% above the upper limit. For content uniformity, it is 70.0%–130.0%. For Category II quantitative tests, the validation range covers 50.0%–120.0% of the acceptance criteria.
accuracy
The accuracy of a quantitative NMR analytical procedure should be determined across the required analytical range. Typically, three levels of concentrations are evaluated using triplicate preparations at each level.
For drug substance assays (Category I), accuracy can be determined by analyzing a reference standard of known purity. For drug product (Category I), a composite sample of reference standard and other components in a pharmaceutical finished product should be used for analytical procedure validation. The assay results are compared to the theoretical value of the reference standard to estimate errors or percent recovery. For the quantitation of impurities (Category II), the accuracy of the analytical procedure can be determined by conducting studies with drug substances or products spiked with known concentrations of the analyte under test. It is also acceptable to compare assay results from the analytical procedure being validated to those of an established, alternative analytical procedure.
Validation criteria:
98.0%–102.0% recovery for drug substances, 95.0%–105.0% recovery for compounded pharmaceutical finished products assay, and 80.0%–120.0% recovery for the quantitative impurity analysis. These criteria should be met throughout the intended range.
precision
Repeatability:
The analytical procedure should be assessed by measuring the concentrations of six separate standard solutions at 100% of the test concentration. The relative standard deviation from the replicate measurements should be evaluated to meet acceptance criteria. Alternatively they can measure the concentrations of three replicates of three separate sample solutions at different concentrations. The three concentrations should be close enough so that the repeatability is constant across the concentration range. If this is done, the repeatability at the three concentrations is pooled for comparison to the acceptance criteria.
Validation criteria:
The relative standard deviation is NMT 1.0% for drug substances, NMT 2.0% for compounded pharmaceutical finished products, and NMT 20.0% for the quantitative impurity analysis.
Intermediate precision:
The effect of random events on the analytical precision of the analytical procedure should be established. Typical variables include performing the analysis on different days, using different instrumentation that are suitable as specified in the analytical procedure, and/or having the analytical procedure performed by two or more analysts. As a minimum, any combination of at least two of these factors totaling six experiments will provide an estimation of intermediate precision.
Validation criteria:
The relative standard deviation is NMT 1.0% for drug substances, NMT 3.0% for compounded pharmaceutical finished products, and NMT 25.0% for quantitative impurity analysis.
quantitation limit (ql)
The QL can be validated by measuring six replicates of test samples spiked with analyte at 50% of specification.
From these replicates, accuracy and precision can be determined. Examples of specifications for Category II quantitative determinations are that the measured concentration is within 70.0%–130.0% of the spike concentration and the relative standard deviation is NMT 15%.
robustness
The reliability of an analytical measurement should be demonstrated with deliberate changes to critical experimental parameters. This can include measuring the stability of the analyte under specified storage conditions, slightly varied inter-pulse delay, probe temperature, and possible interfering species, to list a few examples. Robustness is required for Category I and Category II, quantitative methods.
Analytical Procedure Verification
U.S. Current Good Manufacturing Practices regulations [21 CFR 211.194(a)(2)] indicate that users of analytical procedures described in USP–NF do not need to validate these procedures if provided in a monograph. Instead, they must simply verify their suitability under actual conditions of use.
The objective of an NMR procedure verification is to demonstrate that the procedure as prescribed in a specific monograph can be executed by the user with suitable accuracy, specificity, and precision using the instruments, analysts, and sample matrices available. According to general information chapter Verification of Compendial Procedures 1226, if the verification of the compendial procedure by following the monograph is not successful, the procedure may not be suitable for use with the article under test. It may be necessary to develop and validate an alternative procedure as allowed in General Notices and Requirements 6.30.
Verification of a compendial NMR procedure should at minimum include the execution of the validation parameters for specificity, accuracy, precision, and limit of quantitation, when appropriate, as indicated in this section.
GLOSSARY
Internal standard: An internal standard (IS) is a substance added to a sample solution at a known concentration. One should select an IS with at least a single NMR resonance that does not overlap with those of the analyte. The ratio of a specific internal standard peak area and that of an analyte peak area is used to determine the concentration of the analyte. The number of nuclei corresponding to the integrated peaks in the IS and analyte spectra must be known.
NMR reference: An NMR reference, also known as an NMR shift reference, is a substance added to a sample and from which the chemical shift for the
Reference standard: A reference standard is a substance authenticated by appropriate experimental means to be of a specific chemical structure. In NMR spectroscopy, a reference standard is typically used for the qualitative analysis of a test material. Structure can be confirmed if one directly compares the chemical shifts and multiplicities of the peaks in the NMR spectrum of the test material against the spectrum of the reference standard acquired under comparable experimental conditions.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Kahkashan Zaidi, Ph.D.
Senior Scientific Liaison (301) 816-8269 |
(GCCA2010) General Chapters - Chemical Analysis |
USP38–NF33 Page 528
Pharmacopeial Forum: Volume No. 37(6)