



Add the following:
A shipping container that contains a single article, unless the container also is essentially the immediate container or the outside of the consumer package, must be labeled with a minimum of product identification (except for controlled articles), lot number, expiration date, and conditions for storage and distribution.
Exceptions:
Labeling:
-
Name of the preparation
-
In the case of a liquid preparation, the quantity or proportion of each active moiety and/or drug substance in a specified volume
-
In the case of a compounded sterile preparation, the names and amounts or concentrations of active moiety and/or drug substance on the immediate container (see Pharmaceutical Compounding—Sterile Preparations
 797
797 , Responsibility of Compounding Personnel)
, Responsibility of Compounding Personnel)
-
In the case of a dry preparation or other preparation to which a diluent must be added before use, the quantity or proportion of each active moiety and/or drug substance, the final volume of solution or suspension, directions for proper storage of the constituted solution, and an expiration or beyond-use date (see the section Expiration Date and Beyond-Use Date)
-
Route(s) of administration
-
Name and proportion of all inactive ingredients except ingredients added to adjust the pH or to make the drug isotonic may be declared by name with a statement of their effect
-
Statement of storage conditions
-
Name and place of business of the manufacturer, or packer, or distributor
-
Identifying lot number and expiration date
-
“Rx only”
-
The recommended or usual dosage.
The container shall be labeled so that a sufficient area of the container remains uncovered for its full length or circumference to permit inspection of the contents.
The lot number must be traceable to the complete manufacturing history of the specific package, including all manufacturing, filling, sterilizing, and labeling operations. If the individual monograph permits varying concentrations of active moiety and/or drug substance in a large-volume injection (LVI), the concentration of each active moiety and/or drug substance named in the official title is stated as if it were part of the official title (e.g., 5% Dextrose Injection, or 5% Dextrose and 0.2% Sodium Chloride Injection).
Injections that are intended for veterinary use only should be labeled to that effect.
Vaccine labeling is not included in this general chapter.
Strength and Total Volume for Single- and Multiple-Dose Injectable Drug Products
For single- and multiple-dose injectable drug products, the strength per total volume should be the primary and prominent expression on the principal display panel of the label, followed in close proximity by strength/mL enclosed by parentheses. For containers that hold a volume of less than 1 mL, the strength per fraction of a mL should be the only expression of strength. Strength per single mL should be expressed as mg/mL, not mg/1 mL. The following formats are acceptable for contents greater than 1 mL:
Total strength/total volume: 500 mg/10 mL
Strength/mL: 50 mg/mL
or
Total strength/total volume: 25,000 Units/5 mL
Strength/mL: 5000 Units/mL.
The following format is acceptable for contents less than 1 mL:
12.5 mg/0.625 mL
There are some exceptions to expressing strength per total volume. In certain cases, the primary and prominent expression of the total drug content per container would not be effective in preventing medication errors (e.g., insulin). Another example is the use of lidocaine (or similar drugs for local anesthesia where the product is ordered and administered by percentage (e.g., 1% or 2%). In such cases, the total strength should be expressed: for example, 1% can be expressed as
(100 mg/10 mL) or
(10 mg/mL).
Dry solids that must be reconstituted should follow the same format with the exception that only the total strength of the drug should be listed, not the strength/total volume or strength/mL.
Ratio Expression of Strength
Single-entity drug products that can also be expressed as a ratio, such as epinephrine, shall be labeled only in terms of strength/mL. A ratio expression such as 1:1000 is an unacceptable format for single-entity drug products.
Examples:
Epinephrine Injection, USP, 1:1000 shall be expressed as 1 mg/mL
Epinephrine Injection, USP, 1:10,000 shall be expressed as 0.1 mg/mL
Isoproterenol Hydrochloride Injection, USP, 1:5000 shall be expressed as 0.2 mg/mL
Neostigmine Methylsulfate Injection, 1:1000 shall be expressed as 1 mg/mL
When combined with a local anesthetic, the concentration of epinephrine will be expressed as a ratio.
Examples:
Lidocaine HCl 1% and Epinephrine 1:100,000 Injection, USP
Bupivacaine HCl 0.25% and Epinephrine 1:200,000 Injection, USP
Pharmacy Bulk Package
Where a container is offered as a Pharmacy Bulk Package, the label shall: (a) state prominently “Pharmacy Bulk Package—Not for direct infusion”; (b) contain or refer to information on proper techniques to help assure safe use of the product; and (c) bear a statement limiting the time frame in which the container may be used once it has been entered, provided it is held under labeled storage conditions (see Packaging and Storage Requirements 659).
Ferrules and Cap Overseals
Healthcare practitioners using injectable products must be able to easily see and act on labeling statements that convey important safety messages critical for the prevention of imminent life-threatening situations. These cautionary labeling statements must be simple, concise, and devoid of nonessential information. Products that do not require cautionary statements should be free of information, so that those with cautionary statements are immediately apparent. Accomplishing this requires a systematic approach to the labeling of injectable products, and one that ensures that the ferrule and cap overseal—an area of these products that is highly visible to practitioners as they use these medicines—is reserved for critical safety messages. Accordingly:
-
Only cautionary statements may appear on the top (circle) surface of the ferrule and cap overseal of a vial containing an injectable product. The cautionary statement should appear on both the ferrule and cap, but may appear solely on the ferrule if the cap overseal is transparent and the cautionary statement beneath the cap is readily legible. A cautionary statement is one intended to prevent an imminent life-threatening situation and may include instructional statements that provide potency or other safety-related instructions if warranted. Examples of such statements include, but are not limited to: “Warning—Paralyzing Agent” and “Dilute before Using.” The cautionary statement should be printed in a contrasting color and should be clearly visible under ordinary conditions of use.
-
If no cautionary statement is necessary, the top surface of the vial, including the ferrule and cap overseal, must remain blank.
-
Other statements or features including, but not limited to, identifying numbers or letters, such as code numbers, lot numbers, company names, logos, or product names, etc., may appear on the side (skirt) surface of the ferrule on vials containing injectable products, but not on the top (circle) surface of the ferrule or cap overseal. The appearance of such statements or features on the skirt surface of the ferrule should not detract from, or interfere with, the cautionary statement on the top surface.
Potassium Chloride for Injection Concentrate
The use of a black closure system on a vial (e.g., a black cap overseal and a black ferrule to hold the elastomeric closure) or the use of a black band or series of bands above the constriction on an ampul is prohibited, except for Potassium Chloride for Injection Concentrate (see Packaging and Storage Requirements 659).
Neuromuscular Blocking and Paralyzing Agents
All injectable preparations of neuromuscular blocking agents and paralyzing agents must be packaged in vials with a cautionary statement printed on the ferrules and cap overseals. Both the container cap ferrule and the cap overseal must bear in black or white print (whichever provides the greatest color contrast with the ferrule or cap color) the words: “Warning: Paralyzing Agent” or “Paralyzing Agent” (depending on the size of the closure system). Alternatively, the overseal may be transparent and without words, allowing for visualization of the warning labeling on the closure ferrule.
Aluminum in Large-Volume Injections (LVIs), Small-Volume Injections (SVIs), and Pharmacy Bulk Packages (PBPs) Used in Total Parenteral Nutrition Therapy
-
The aluminum content of LVIs used in total parenteral nutrition (TPN) therapy must not exceed 25 mcg/L.
-
The package insert of LVIs used in TPN therapy must state that the drug product contains NMT 25 mcg of aluminum per L. This information must be contained in the Precautions section of the labeling of all LVIs used in TPN therapy.
-
If the maximum amount of aluminum in SVIs and PBPs is 25 mcg/L or less, instead of stating the exact amount of aluminum that each contains, as in paragraph (4), the immediate container label for SVIs and PBPs used in the preparation of TPN admixtures or formulations (with exceptions as noted below) may state: “Contains no more than 25 mcg/L of aluminum.” If the SVI or PBP is a lyophilized powder, the immediate container label may state the following: “When reconstituted in accordance with the package insert instructions, the concentration of aluminum will be no more than 25 mcg/L.”
-
The maximum level of aluminum at expiry must be stated on the immediate container label of all SVIs and PBPs used in the preparation of TPN admixtures or formulations. The aluminum content must be stated as follows: “Contains no more than ___ mcg/L of aluminum.” The immediate container label of all SVIs and PBPs that are lyophilized powder used in the preparation of TPN solutions must contain the following statement: “When reconstituted in accordance with the package insert instructions, the concentration of aluminum will be no more than ___ mcg/L.” This maximum amount of aluminum must be stated as the highest one of the following three levels:
-
The highest level for the batches produced during the past 3 years
-
The highest level for the latest five batches
-
The maximum level in terms of historical levels, but only until completion of production of the first five batches.
-
The highest level for the batches produced during the past 3 years
The package insert for all LVIs, SVIs, and PBPs used in the preparation of TPN admixtures or formulations shall contain the following statement in the Warnings section of the labeling:
WARNING: This product contains aluminum which may be toxic. Aluminum may reach toxic levels with prolonged parenteral administration if kidney function is impaired. Premature neonates are particularly at risk because of their kidneys, which are immature, and they require large amounts of calcium and phosphate solutions that contain aluminum.
Research indicates that patients with impaired kidney function, including premature neonates, who receive parenteral levels of aluminum at greater than 4–5 mcg/kg/day, accumulate aluminum at levels associated with central nervous system and bone toxicity. Tissue loading may occur at even lower rates of administration.
-
In the case of a liquid preparation, the percentage content of each active moiety and/or drug substance or the amount of each active moiety and/or drug substance in a specified volume, with the exception that the ingedients added to adjust to a given pH or to make the solution isotonic may be declared by name and a statement of their effect.
-
In the case of a compounded preparation, the labeling should indicate that “this is a compounded preparation” (see Pharmaceutical Compounding—Nonsterile Preparations 795, Compounding Process, Criteria When Compounding Each Drug Preparation).
-
In the case of a dry preparation or other preparation to which a diluent must be added before use, the amount of each active moiety and/or drug substance, the composition of recommended diluent(s) [the name(s) alone if the formula is specified in the individual monograph], the amount that will be used to attain a specific concentration of active moiety or drug substance, the final volume of solution, directions for proper storage of the constituted solution, and an expiration or beyond-use date (see the section Expiration Date and Beyond-Use Date).
Amount of Active Moiety and/or Drug Substance per Dosage Unit
The strength of a drug product is expressed on the container label in terms of micrograms, milligrams, grams, or percentage of the therapeutically active moiety or drug substance, whichever form is used in the title, unless otherwise indicated in an individual monograph. Both the active moiety and drug substance names and their equivalent amounts are then provided on the container label and in the labeling (see Nomenclature 1121, Monograph Naming Policy for Salt Drug Substances in Drug Products and Compounded Preparations).
Official articles in capsule, tablet, or other dosage forms shall be labeled to express the quantity of each active moiety and/or drug substance or recognized nutrient contained in each unit. Unit-dose oral solutions or suspensions (whether supplied as liquid preparations or as liquid preparations that are constituted from solids upon addition of a designated volume of a specific diluent) shall be labeled to express the quantity of each active moiety and/or drug substance or recognized nutrient delivered under the conditions prescribed in Deliverable Volume 698. Official drug products not in unit-dose packaging shall be labeled to show the quantity of each active moiety and/or drug substance in each milliliter or in each gram, or to express the percentage of each such ingredient (see General Notices and Requirements 8.140, Percentage Concentrations). Exceptions are oral liquids or solids intended to be constituted to yield oral liquids that, alternatively, can be labeled in terms of each 5-mL portion of the liquid or resulting liquid. Unless otherwise indicated in a monograph or chapter, declarations of strength or quantity shall be stated only in metric units [see also General Notices and Requirements 5.50.10, Units of Potency (Biological)].
Expiration Date and Beyond-Use Date
The label of an official drug product or nutritional or dietary supplement product shall bear an expiration date. All products shall display the expiration date so that it can be read by an ordinary individual under customary conditions of purchase and use. The expiration date shall be prominently displayed in high contrast to the background or it shall be sharply embossed, and easily understood (e.g., “EXP 6/13,” “Exp. June 13,” or “Expires 6/2013”). [Note—For additional information and guidance, refer to the Consumer Products Association's Voluntary Codes and Guidelines of the Consumer Healthcare Products Industry.
The monographs for some preparations state how the labeled expiration date shall be determined. In the absence of a specific requirement in the individual monograph for a drug product or nutritional supplement, the label shall bear an expiration date assigned for the particular formulation and package of the product, with the following exceptions: the label need not show an expiration date if the drug product or nutritional supplement is packaged in a container that is intended for sale without prescription, and the labeling states no dosage limitations, and if the product or supplement is stable for NLT 3 years when stored under the prescribed conditions.
If an official product is required to bear an expiration date, the product shall be dispensed solely in or from a container labeled with an expiration date, and the date on which the article is dispensed shall be within the labeled expiry period. The expiration date identifies the time during which the article can be expected to meet the requirements of the compendial monograph, provided it is kept under the prescribed storage conditions. The expiration date limits the time during which the article may be dispensed or used. If an expiration date is stated only in terms of the month and the year, then the intended expiration date is the last day of the stated month.
The beyond-use date is the date after which a product shall not be used. The dispenser shall place on the label of the prescription container a suitable beyond-use date to limit the patient's use of the article based on any information supplied by the manufacturer or this subsection. The beyond-use date shall not be later than the expiration date on the manufacturer's container. Also see the section Compounded Preparations below.
For articles that require constitution before use, a suitable beyond-use date for the constituted product shall be identified in the labeling.
For all other dosage forms, in determining a beyond-use date the dispenser shall take into account, in addition to any other relevant factors:
-
Nature of the drug
-
Container in which it was packaged by the manufacturer and the expiration date thereon
-
Characteristics of the patient's container, if the article is repackaged for dispensing
-
Expected storage conditions to which the article may be exposed
-
Unusual storage conditions to which the article may be exposed
-
Expected length of the course of therapy.
After considering these factors, the dispenser shall label a container with a suitable beyond-use date to limit the patient's use of the article. Unless otherwise specified in the individual monograph or in the absence of stability data to the contrary, the beyond-use date shall be not later than: (a) the expiration date on the manufacturer's container; or (b) 1 year from the date the drug is dispensed, whichever is earlier. For nonsterile solid and liquid dosage forms that are packaged in single-unit and unit-dose containers, the beyond-use date shall be 1 year from the date the drug is packaged into the single-unit or unit-dose container or the expiration date on the manufacturer's container, whichever is earlier, unless stability data or the manufacturer's labeling indicates otherwise.
Compounded Preparations
The label on the container or package of an official compounded preparation shall bear a beyond-use date after which the compounded preparation should not be used. Beyond-use dates are assigned on the basis of criteria different from those applied to assigning expiration dates to manufactured drug products.
The label on the container package of an official compounded preparation shall include the word “compounded” after the drug name (e.g., Baclofen Compounded Oral Solution). Additionally, USP official compounded preparations for animal patients will include the word “veterinary” following the full official name (e.g., Atenolol Compounded Suspension, Veterinary).
Irrigation, Hemofiltration, and Dialysis
Flexible containers for injections that are intended for use as dialysis, hemofiltration, or irrigation solutions, and that contain a volume of more than 1 L, should be labeled to indicate that the contents are not intended for use by intravenous infusion.
Use of Leading and Terminal Zeros
To help minimize the possibility of errors in drug dispensing and administration, when the quantity of active moiety and/or drug substance is expressed in whole numbers it shall be shown without a decimal point followed by a terminal zero (e.g., express as 4 mg, not 4.0 mg). When the quantity of active moiety and/or drug substance is expressed as a decimal number smaller than 1, it shall be shown with a zero preceding the decimal point (e.g., express as 0.2 mg, not .2 mg).
Alcohol
The alcohol content in a liquid preparation shall be stated on the label as a percentage (v/v) of C2H5OH.
Botanicals
The label of an herb or other botanical intended for use as a dietary supplement shall bear the statement, “If you are pregnant or nursing a baby, seek the advice of a health professional before using this product.”
Electrolytes
The concentration of electrolytes for replacement therapy (e.g., sodium, potassium, chloride) shall be stated on the label in milliequivalents (mEq)/volume. [Note—Phosphorus containing injections shall be expressed in milliMoles (e.g., mM/volume). The label of the product shall also indicate the quantity of ingredient(s) in terms of weight or percentage concentration.
Non-Oral Products
A product intended for injection or topical use shall state the names of all added substances (see General Notices and Requirements 5.20, Added Substances).
Salts of Drugs
It is an established principle that official articles shall have only one official title (see General Notices and Requirements 2.20 and compendial nomenclature requirements in Nomenclature 1121). For purposes of saving space on labels and because chemical symbols for the most common inorganic salts of drugs are well known to practitioners, the following alternatives are permitted in labeling official articles that are salts: HCl for hydrochloride; HBr for hydrobromide; Na for sodium; and K for potassium. The symbols Na and K are intended for use in abbreviating names of the salts of organic acids, but these symbols are not used when the word Sodium or Potassium appears at the beginning of an official title (e.g., Phenobarbital Na is acceptable, but Na Salicylate is not).
Special Capsules and Tablets
The label of any form of Capsule or Tablet intended for administration other than by swallowing intact shall bear a prominent indication of the manner in which it should be used (see Compendial Nomenclature, Nomenclature Guidelines on the USP website at www.usp.org).
Products That Contain Vitamins
The vitamin content of an official drug product shall be stated on the label in metric units per dosage unit. The amounts of vitamins A, D, and E may also be stated in USP Units. Quantities of vitamin A declared in metric units refer to the equivalent amounts of retinol (vitamin A alcohol). The label of a nutritional supplement shall bear an identifying lot number, control number, or batch number.
Controlled Room Temperature
Articles may be labeled for storage at “controlled room temperature” or at “up to 25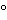 ”, or other wording based on the same mean kinetic temperature (see Packaging and Storage Requirements 659).
”, or other wording based on the same mean kinetic temperature (see Packaging and Storage Requirements 659).
Light-Resistant Container
When an opaque covering is used to provide protection from light for a light-sensitive product packaged in a clear or colorless or translucent container, the label of the container will bear a statement that the opaque covering is needed until the contents are to be used or administered (see Containers—Performance Testing 671, Light Transmission Test and Packaging and Storage Requirements 659).
Single-Unit Container
Each single-unit container shall be labeled to indicate the identity, quantity and/or strength, name of the manufacturer, lot number, and expiration date of the article (see Packaging and Storage Requirements 659).
Single-Dose Container
A single-dose container shall be labeled as such, and when space permits, should include on the label appropriate discard instructions (see Packaging and Storage Requirements 659).
Unit-of-Use Container
A unit-of-use container shall be labeled as such, without further modification except for the addition of appropriate labeling (see Packaging and Storage Requirements 659).
Protection from Freezing
The container label shall bear an appropriate instruction to protect the article from freezing if subject to loss of strength or potency, or to destructive alteration of its characteristics (see Packaging and Storage Requirements 659).
Prescription Container Labeling
At a minimum, a prescription container shall be labeled in a patient-centered manner. The label shall contain essential information that is important for the patient's safe and effective use of the medicine. Labels should be designed and formatted to optimize readability and understanding (see Prescription Container Labeling 17).
Official May 1, 2016
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Donna M. Bohannon, R.Ph.
Scientific Liaison, Nomenclature, Safety and Labeling (301) 230-3252 |
(NSL2010) Nomenclature Safety and Labeling |
USP38–NF33 Page 87
Pharmacopeial Forum: Volume No. 39(6)