The following definitions and general procedures apply to fats, fixed oils, waxes, resins, balsams, and similar substances.
PREPARATION OF SPECIMEN
If a specimen of oil shows turbidity owing to separated stearin, warm the container in a water bath at 50 until the oil is clear, or if the oil does not become clear on warming, pass it through dry filter paper in a funnel contained in a hot-water jacket. Mix thoroughly, and weigh at one time as many portions as are needed for the various determinations, using preferably a bottle having a pipet dropper, or a weighing buret. Keep the specimen melted, if solid at room temperature, until the desired portions of specimen are withdrawn.
until the oil is clear, or if the oil does not become clear on warming, pass it through dry filter paper in a funnel contained in a hot-water jacket. Mix thoroughly, and weigh at one time as many portions as are needed for the various determinations, using preferably a bottle having a pipet dropper, or a weighing buret. Keep the specimen melted, if solid at room temperature, until the desired portions of specimen are withdrawn.
SPECIFIC GRAVITY
Determine the specific gravity of a fat or oil as directed under Specific Gravity  841
841 .
.
MELTING TEMPERATURE
Determine the melting temperature as directed for substances of Class II (see Melting Range or Temperature 741).
ACID VALUE (FREE FATTY ACIDS)
The acidity of fats and fixed oils in this Pharmacopeia may be expressed as the number of mL of 0.1 N alkali required to neutralize the free acids in 10.0 g of substance. Acidity is frequently expressed as the Acid Value, which is the number of mg of potassium hydroxide required to neutralize the free acids in 1.0 g of the substance. Unless otherwise directed in the individual monograph, use Method I.
Method I
Procedure—
Unless otherwise directed, dissolve about 10.0 g of the substance, accurately weighed, in 50 mL of a mixture of equal volumes of alcohol and ether (which has been neutralized to phenolphthalein with 0.1 N potassium hydroxide or 0.1 N sodium hydroxide, unless otherwise specified) contained in a flask. If the test specimen does not dissolve in the cold solvent, connect the flask with a suitable condenser and warm slowly, with frequent shaking, until the specimen dissolves. Add 1 mL of phenolphthalein TS, and titrate with 0.1 N potassium hydroxide VS or 0.1 N sodium hydroxide VS until the solution remains faintly pink after shaking for 30 seconds. Calculate either the Acid Value or the volume of 0.1 N alkali required to neutralize 10.0 g of specimen (free fatty acids), whichever is appropriate. Calculate the Acid Value by the formula:
56.11V × N/W
in which 56.11 is the molecular weight of potassium hydroxide; V is the volume, in mL; N is the normality of the potassium hydroxide solution or the sodium hydroxide solution; and W is the weight, in g, of the sample taken.
If the volume of 0.1 N potassium hydroxide VS or 0.1 N sodium hydroxide VS required for the titration is less than 2 mL, a more dilute titrant may be used, or the sample size may be adjusted accordingly. The results may be expressed in terms of the volume of titrant used or in terms of the equivalent volume of 0.1 N potassium hydroxide or 0.1 N sodium hydroxide.
If the oil has been saturated with carbon dioxide for the purpose of preservation, gently reflux the alcohol-ether solution for 10 minutes before titration. The oil may be freed from carbon dioxide also by exposing it in a shallow dish in a vacuum desiccator for 24 hours before weighing the test specimens.
Method II
Procedure—
Prepare 125 mL of a solvent mixture consisting of equal volumes of isopropyl alcohol and toluene. Before use, add 2 mL of a 1% solution of phenolphthalein in isopropyl alcohol to the 125-mL mixture, and neutralize with alkali to a faint but permanent pink color. Weigh accurately the appropriate amount of well-mixed liquid sample indicated in the table below, and dissolve it in the neutralized solvent mixture. If the test specimen does not dissolve in the cold solvent, connect the flask with a suitable condenser and warm slowly, with frequent shaking, until the specimen dissolves. Shake vigorously while titrating with 0.1 N potassium hydroxide VS or 0.1 N sodium hydroxide VS to the first permanent pink of the same intensity as that of the neutralized solvent before mixing with the sample. Calculate the Acid Value as indicated in Method I.
| Acid Value | Sample Weight (g) |
|---|---|
| 0–1 | 20 |
| 1–4 | 10 |
| 4–15 | 2.5 |
| 15–74.9 | 0.5 |
| 0.1 |
Change to read:
ESTER VALUE
The Ester Value is the number of mg of potassium hydroxide required to saponify the esters in 1.0 g of the substance. If the Saponification Value and the Acid Value have been determined, the difference between these two represents the Ester Value, i.e., Ester Value = Saponification Value – Acid Value.
Procedure—
Place 1.5–2 g of the substance, accurately weighed, in a tared, 250-mL flask, add 20–30 mL of neutralized alcohol, and shake. Add 1 mL of phenolphthalein TS, and titrate with 0.5 N alcoholic potassium hydroxide VS until the free acid is neutralized. Add 25.0 mL of 0.5 N alcoholic potassium hydroxide VS, and proceed as directed under Saponification Value, beginning with “Heat the flask” and omitting the further addition of phenolphthalein TS. Calculate the Ester Value by the formula:
 [56.11(VB
[56.11(VB  VT)N]/WUSP35
in which 56.11 is the molecular weight of potassium hydroxide; VB and VT are the volumes, in mL, of 0.5 N hydrochloric acid consumed in the blank test and in the actual test, respectively; N is the exact normality of the hydrochloric acid; and W is the weight, in g, of the substance taken for the test.
VT)N]/WUSP35
in which 56.11 is the molecular weight of potassium hydroxide; VB and VT are the volumes, in mL, of 0.5 N hydrochloric acid consumed in the blank test and in the actual test, respectively; N is the exact normality of the hydrochloric acid; and W is the weight, in g, of the substance taken for the test.
HYDROXYL VALUE
The Hydroxyl Value is the number of mg of potassium hydroxide equivalent to the hydroxyl content of 1.0 g of the substance.
Pyridine–Acetic Anhydride Reagent—
Just before use, mix 3 volumes of freshly opened or freshly distilled pyridine with 1 volume of freshly opened or freshly distilled acetic anhydride.
Procedure—
Transfer a quantity of the substance, determined by reference to the accompanying table and accurately weighed, to a glass-stoppered, 250-mL conical flask, and add 5.0 mL of Pyridine–Acetic Anhydride Reagent. Transfer 5.0 mL of Pyridine–Acetic Anhydride Reagent to a second glass-stoppered, 250-mL conical flask to provide the reagent blank. Fit both flasks with suitable glass-jointed reflux condensers, heat on a steam bath for 1 hour, add 10 mL of water through each condenser, and heat on the steam bath for 10 minutes more. Cool, and to each add 25 mL of butyl alcohol, previously neutralized to phenolphthalein TS with 0.5 N alcoholic potassium hydroxide, by pouring 15 mL through each condenser and, after removing the condensers, washing the sides of both flasks with the remaining 10-mL portions. To each flask add 1 mL of phenolphthalein TS, and titrate with 0.5 N alcoholic potassium hydroxide VS, recording the volume, in mL, consumed by the residual acid in the test solution as T and that consumed by the blank as B. In a 125-mL conical flask, mix about 10 g of the substance, accurately weighed, with 10 mL of freshly distilled pyridine, previously neutralized to phenolphthalein TS, add 1 mL of phenolphthalein TS, and titrate with 0.5 N alcoholic potassium hydroxide VS, recording the volume, in mL, consumed by the free acid in the test specimen as A. Calculate the Hydroxyl Value by the formula:
(56.11N/W)[B + (WA/C) T]
in which W and C are the weights, in g, of the substances taken for the acetylation and for the free acid determination, respectively; N is the exact normality of the alcoholic potassium hydroxide; and 56.11 is the molecular weight of potassium hydroxide. If the Acid Value for the test substance is known, calculate the Hydroxyl Value by the formula:
(56.11N/W)[B – T] + Acid Value
in which W is the weight, in g, of the substance taken for the acetylation; N is the exact normality of the alcoholic potassium hydroxide; and 56.11 is the molecular weight of potassium hydroxide.
| Hydroxyl Value Range |
Weight of Test Specimen (g) |
|---|---|
| 0–20 | 10 |
| 20–50 | 5 |
| 50–100 | 3 |
| 100–150 | 2 |
| 150–200 | 1.5 |
| 200–250 | 1.25 |
| 250–300 | 1.0 |
| 300–350 | 0.75 |
IODINE VALUE
The Iodine Value represents the number of g of iodine absorbed, under the prescribed conditions, by 100 g of the substance. Unless otherwise specified in the individual monograph, determine the Iodine Value by Method I.
Method I (Hanus Method)
Procedure—
Transfer an accurately weighed quantity of sample, as determined from the accompanying table, into a 250-mL iodine flask, dissolve it in 10 mL of chloroform, add 25.0 mL of iodobromide TS, insert the stopper in the vessel securely, and allow it to stand for 30 minutes protected from light, with occasional shaking. Then add, in the order named, 30 mL of potassium iodide TS and 100 mL of water, and titrate the liberated iodine with 0.1 N sodium thiosulfate VS, shaking thoroughly after each addition of thiosulfate. When the iodine color becomes quite pale, add 3 mL of starch TS, and continue the titration with 0.1 N sodium thiosulfate VS until the blue color is discharged. Perform a blank test at the same time with the same quantities of the same reagents and in the same manner (see Residual Titrations 541). Calculate the Iodine Value from the formula:
[126.90(VB VS)N]/(10W)
in which 126.90 is the atomic weight of iodine; VB and VS are the volumes, in mL, of 0.1 N sodium thiosulfate VS consumed by the blank test and the actual test, respectively; N is the exact normality of the sodium thiosulfate VS; and W is the weight, in g, of the substance taken for the test.
[Note—If more than half of the iodobromide TS is absorbed by the portion of the substance taken, repeat the determination, using a smaller portion of the substance under examination. ]
Sample Weights
| Iodine Value Expected |
Weight in g, ±0.1 |
|---|---|
| <5 | 3.0 |
| 5–20 | 1.0 |
| 21–50 | 0.4 |
| 51–100 | 0.2 |
| 101–150 | 0.13 |
| 151–200 | 0.1 |
Method II
Potassium Iodide Solution—
Dissolve 10.0 g of potassium iodide in water to make 100 mL. Store in light-resistant containers.
Starch Indicator Solution—
Mix 1 g of soluble starch with sufficient cold water to make a thin paste. Add, while stirring, to 100 mL of boiling water. Mix, and cool. Use only the clear solution.
Procedure—
Melt the sample, if it is not already liquid. [Note—The temperature during melting should not exceed the melting point of the sample by more than 10. ] Pass through two pieces of filter paper to remove any solid impurities and the last traces of moisture. The filtration may be performed in an air oven at 100 but should be completed within 5 minutes ± 30 seconds. The sample must be absolutely dry. All glassware must be absolutely clean and completely dry. After filtration, allow the filtered sample to achieve a temperature of 68 to 71 ± 1 before weighing the sample. Once the sample has achieved a temperature of 68 to 71 ± 1, immediately weigh the sample into a 500-mL iodine flask, using the weights and weighing accuracy noted in the accompanying table. [Note—The weight of the substance must be such that there will be an excess of iodochloride TS of 50%–60% of the amount added, that is, 100%–150% of the amount absorbed. ] Add 15 mL of a fresh mixture of cyclohexane and glacial acetic acid (1:1), and swirl to dissolve the sample. Add 25.0 mL of iodochloride TS, insert the stopper securely in the flask, and swirl to mix. Allow it to stand at 25 ± 5, protected from light, with occasional shaking, for 1.0 or 2.0 hours, depending on the Iodine Value (IV) of the sample: IV less than 150, 1.0 hour; IV equal to or greater than 150, 2.0 hours. Then, within 3 minutes after the indicated reaction time, add, in the order named, 20 mL of Potassium Iodide Solution and 150 mL of recently boiled and cooled water, and mix. Within 30 minutes, titrate the liberated iodine with 0.1 N sodium thiosulfate VS, while stirring by mechanical means after each addition of thiosulfate. When the yellow iodine color has almost disappeared, add 1–2 mL of Starch Indicator Solution, and continue the titration with 0.1 N sodium thiosulfate VS until the blue color is discharged. Perform a blank test at the same time with the same quantities of the same reagents and in the same manner (see Residual Titrations 541). Calculate the Iodine Value as indicated in Method I.
PEROXIDE VALUE
The Peroxide Value is the number that expresses, in milliequivalents of active oxygen, the quantity of peroxide contained in 1000 g of the substance. [Note—This test must be performed promptly after sampling to avoid oxidation of the test specimen. ]
Procedure—
Unless otherwise directed, place about 5 g of the substance, accurately weighed, in a 250-mL conical flask fitted with a ground-glass stopper. Add 30 mL of a mixture of glacial acetic acid and chloroform (3:2), shake to dissolve, and add 0.5 mL of saturated potassium iodide solution. Shake for exactly 1 minute, and add 30 mL of water. Titrate with 0.01 N sodium thiosulfate VS, adding the titrant slowly with continuous shaking, until the yellow color is almost discharged. Add 5 mL of starch TS, and continue the titration, shaking vigorously, until the blue color is discharged. Perform a blank determination under the same conditions. [Note—The volume of titrant used in the blank determination must not exceed 0.1 mL. ] Calculate the Peroxide Value by the formula:
[1000 (VT – VB)N]/W
in which VT and VB are the volumes, in mL, of 0.01 N sodium thiosulfate consumed in the actual test and in the blank test, respectively; N is the exact normality of the sodium thiosulfate solution; and W is the weight, in g, of the substance taken for the test.
SAPONIFICATION VALUE
The Saponification Value is the number of mg of potassium hydroxide required to neutralize the free acids and saponify the esters contained in 1.0 g of the substance.
Procedure—
Place 1.5–2 g of the substance in a tared, 250-mL flask, weigh accurately, and add to it 25.0 mL of 0.5 N alcoholic potassium hydroxide. Heat the flask on a steam bath, under a suitable condenser to maintain reflux for 30 minutes, frequently rotating the contents. [Note—Reflux time can be up to 90 minutes to ensure complete saponification, depending on the type of ester to be tested. ] Then add 1 mL of phenolphthalein TS, and titrate the excess potassium hydroxide with 0.5 N hydrochloric acid VS. Perform a blank determination under the same conditions (see Residual Titrations under Titrimetry 541). The titration also can be carried out potentiometrically. Calculate the Saponification Value by the formula:
[56.11(VB – VT)N]/W
in which 56.11 is the molecular weight of potassium hydroxide; VB and VT are the volumes, in mL, of 0.5 N hydrochloric acid consumed in the blank test and in the actual test, respectively; N is the exact normality of the hydrochloric acid; and W is the weight, in g, of the substance taken for the test.
If the oil has been saturated with carbon dioxide for the purpose of preservation, expose it in a shallow dish in a vacuum desiccator for 24 hours before weighing the test specimens.
UNSAPONIFIABLE MATTER
The term “Unsaponifiable Matter” in oils or fats, refers to those substances that are not saponifiable by alkali hydroxides but are soluble in the ordinary fat solvents, and to products of saponification that are soluble in such solvents.
Procedure—
Transfer about 5.0 g of the oil or fat, accurately weighed, to a 250-mL conical flask, add 50 mL of an alcoholic potassium hydroxide solution prepared by dissolving 12 g of potassium hydroxide in 10 mL of water and diluting this solution with alcohol to 100 mL, and heat the flask on a steam bath under a suitable condenser to maintain reflux for 1 hour, swirling frequently. Cool to a temperature below 25, and transfer the contents of the flask to a separator having a polytetrafluoroethylene stopcock, rinsing the flask with two 50-mL portions of water that are added to the separator (do not use grease on stopcock). Extract with three 100-mL portions of ether, combining the ether extracts in another separator containing 40 mL of water. Gently rotate or shake the separator for a few minutes. [Note—Violent agitation may result in the formation of a difficult-to-separate emulsion. ] Allow the mixture to separate, and discard the lower aqueous phase. Wash the ether extract with two additional 40-mL portions of water, and discard the lower aqueous phase. Wash the ether extract successively with a 40-mL portion of potassium hydroxide solution (3 in 100) and a 40-mL portion of water. Repeat this potassium hydroxide solution–water wash sequence three times. Wash the ether extract with 40-mL portions of water until the last washing is not reddened by the addition of 2 drops of phenolphthalein TS. Transfer the ether extract to a tared flask, and rinse the separator with 10 mL of ether, adding the rinsings to the flask. Evaporate the ether on a steam bath, and add 6 mL of acetone to the residue. Remove the acetone in a current of air, and dry the residue at 105 until successive weighings differ by not more than 1 mg. Calculate the percentage of unsaponifiable matter in the portion of oil or fat taken by the formula:
100(WR/WS)
in which WR is the weight, in g, of the residue; and WS is the weight, in g, of the oil or fat taken for the test.
Dissolve the residue in 20 mL of alcohol, previously neutralized to the phenolphthalein endpoint, add phenolphthalein TS, and titrate with 0.1 N alcoholic sodium hydroxide VS to the first appearance of a faint pink color that persists for not less than 30 seconds. If the volume of 0.1 N alcoholic sodium hydroxide required is greater than 0.2 mL, the separation of the layers was incomplete; the residue weighed cannot be considered as “unsaponifiable matter,” and the test must be repeated.
SOLIDIFICATION TEMPERATURE OF FATTY ACIDS
Preparation of the Fatty Acids—
Heat 75 mL of glycerin–potassium hydroxide solution (made by dissolving 25 g of potassium hydroxide in 100 mL of glycerin) in an 800-mL beaker to 150, and add 50 mL of the clarified fat, melted if necessary. Heat the mixture for 15 minutes with frequent stirring, but do not allow the temperature to rise above 150. Saponification is complete when the mixture is homogeneous, with no particles clinging to the beaker at the meniscus. Pour the contents of the beaker into 500 mL of nearly boiling water in an 800-mL beaker or casserole, add slowly 50 mL of dilute sulfuric acid (made by adding water and sulfuric acid (3:1)), and heat the solution, with frequent stirring, until the fatty acids separate cleanly as a transparent layer. Wash the acids with boiling water until free from sulfuric acid, collect them in a small beaker, place on a steam bath until the water has settled and the fatty acids are clear, filter into a dry beaker while hot, and dry at 105 for 20 minutes. Place the warm fatty acids in a suitable container, and cool in an ice bath until they congeal.
Test for Complete Saponification—
Place 3 mL of the dry acids in a test tube, and add 15 mL of alcohol. Heat the solution to boiling, and add an equal volume of 6 N ammonium hydroxide. A clear solution results.
Procedure—
Using an apparatus similar to the “Congealing Temperature Apparatus” specified therein, proceed as directed for Procedure under Congealing Temperature 651, reading “solidification temperature” for “congealing point” (the terms are synonymous). The average of not less than four consecutive readings of the highest point to which the temperature rises is the solidification temperature of the fatty acids.
Change to read:
FATTY ACID COMPOSITION
Standard Solution—
Prepare an ester mixture of known composition containing the esters required in the individual monograph. This Standard Solution may contain other components. [Note—Ester mixtures are available commercially from Nu-Chek-Prep, Inc., P.O. Box 295, Elysian, MN 56028. Typical Nu-Chek-Prep ester mixtures useful in this test include Nu-Chek 17A and Nu-Chek 19A. ] Nu-Chek mixture 17A has the following composition:
| Percentage | Fatty Acid Ester | Carbon-chain Length | No. of Double Bonds |
|---|---|---|---|
| 1.0 | Methyl myristate | 14 | 0 |
| 4.0 | Methyl palmitate | 16 | 0 |
| 3.0 | Methyl stearate | 18 | 0 |
| 3.0 | Methyl arachidate | 20 | 0 |
| 3.0 | Methyl behenate | 22 | 0 |
| 3.0 | Methyl lignocerate | 24 | 0 |
| 45.0 | Methyl oleate | 18 | 1 |
| 15.0 | Methyl linoleate | 18 | 2 |
| 3.0 | Methyl linolenate | 18 | 3 |
| 20.0 | Methyl erucate | 22 | 1 |
Nu-Chek mixture 19A has the following composition:
| Percentage | Fatty Acid Ester | Carbon-chain Length | No. of Double Bonds |
|---|---|---|---|
| 7.0 | Methyl caprylate | 8 | 0 |
| 5.0 | Methyl caprate | 10 | 0 |
| 48.0 | Methyl laurate | 12 | 0 |
| 15.0 | Methyl myristate | 14 | 0 |
| 7.0 | Methyl palmitate | 16 | 0 |
| 3.0 | Methyl stearate | 18 | 0 |
| 12.0 | Methyl oleate | 18 | 1 |
| 3.0 | Methyl linoleate | 18 | 2 |
Test Solution—
[Note—If fatty acids containing more than 2 double bonds are present in the test specimen, remove air from the flask by purging it with nitrogen for a few minutes. ] Transfer about 100 mg of the test specimen to a 50-mL conical flask fitted with a suitable water-cooled reflux condenser and a magnetic stir bar. Add 4 mL of 0.5 N Methanolic Sodium Hydroxide Solution,USP35 and reflux until fat globules disappear (usually 5–10 minutes). Add 5 mL of a solution prepared by dissolving 14 g of boron trifluoride in methanol to make 100 mL, swirl to mix, and reflux for 2 minutes. Add 4 mL of chromatographic n-heptane through the condenser, and reflux for 1 minute. Cool, remove the condenser, add about 15 mL of saturated sodium chloride solution, shake, and allow the layers to separate. Pass the n-heptane layer through 0.1 g of anhydrous sodium sulfate (previously washed with chromatographic n-heptane) into a suitable flask. Transfer 1.0 mL of this solution to a 10-mL volumetric flask, dilute with chromatographic n-heptane to volume, and mix.
System Suitability Solution—
Transfer about 20 mg each of stearic acid, palmitic acid, and oleic acid to a 25-mL conical flask fitted with a suitable water-cooled reflux condenser and a magnetic stir bar, and proceed as directed for Test Solution, beginning with “Add 5.0 mL of a solution prepared by dissolving.”
Chromatographic System (see Chromatography 621)—
The gas chromatograph is equipped with a flame-ionization detector, maintained at a temperature of about 260, a splitless injection system, and a 0.53-mm × 30-m fused-silica capillary column bonded with a 1.0-µm layer of phase G16. The chromatograph is programmed to maintain the column temperature at 70 for about 2 minutes after injection, then to increase the temperature at the rate of 5 per minute to 240, and finally to maintain this temperature for 5 minutes. The injection port temperature is maintained at about 220. The carrier gas is helium with a linear velocity of about 50 cm per second.
Chromatograph the System Suitability Solution, and record the peak responses as directed for Procedure: the relative retention times are about 0.87 for methyl palmitate, 0.99 for methyl stearate, and 1.0 for methyl oleate; the resolution, R, between methyl stearate and methyl oleate is not less than 1.5; and the relative standard deviation of the peak area responses for the palmitate and stearate peaks for replicate injections is not more than 6.0%. The relative standard deviation of the peak area response ratio of the palmitate to stearate peaks from these replicate injections is not more than 1.0%.
Procedure—
Separately inject equal volumes (about 1 µL) of the Standard Solution and the Test Solution into the chromatograph, record the chromatograms, identify the fatty acid ester peaks in the chromatogram of the Test Solution by comparing the retention times of these peaks with those obtained in the chromatogram of the Standard Solution, and measure the peak areas for all of the fatty acid ester peaks in the chromatogram obtained from the Test Solution. Calculate the percentage of each fatty acid component in the test specimen by the formula:
100(A/B)
in which A is the area of the peak response obtained for each individual fatty acid ester component; and B is the sum of the peak areas of all of the peaks, excluding the solvent peak, in the chromatogram obtained from the Test Solution.
OMEGA-3 FATTY ACIDS DETERMINATION AND PROFILE
The following procedure may be used for the determination of eicosapentaenoic acid (EPA) (C20:5 n-3), docosahexaenoic acid (DHA) (C22:6 n-3) and total omega-3 acids obtained from fish, plant, or microbial sources in bulk oils and encapsulated oil. Protect the solutions from actinic light, oxidizing agents, oxidation catalysts, and air.
Content of EPA and DHA
USP Reference Standards 11—
USP Docosahexaenoic Acid Ethyl Ester RS. USP Eicosapentaenoic Acid Ethyl Ester RS. USP Methyl Tricosanoate RS.
Antioxidant Solution—
Dissolve an accurately weighed quantity of butylated hydroxytoluene in 2,2,4-trimethylpentane to obtain a solution having a concentration of 0.05 mg per mL.
Internal Standard Solution—
Transfer an accurately weighed quantity of USP Methyl Tricosanoate RS to a volumetric flask. Dissolve in Antioxidant Solution, and dilute with the same solvent to obtain a solution having a concentration of about 7.0 mg per mL. [Note—Guard the solution against evaporation during usage. ]
| Approx. Sum EPA + DHA |
Amount of Sample to Be Weighed (g) |
|---|---|
| 30%–50% | 0.4–0.5 |
| 50%–70% | 0.3 |
| 70%–80% | 0.25 |
Test Solution 1 (for triglycerides)—
In a 10-mL volumetric flask, dissolve the mass of sample to be examined, according to the table above, in Antioxidant Solution, and dilute with the same solution to volume. Transfer 2.0 mL of this solution to a glass tube, and evaporate the solvent with a gentle stream of nitrogen. Add 1.5 mL of a 2% (w/v) solution of sodium hydroxide in methanol, cap tightly with a polytetrafluoroethylene-lined cap, mix, and heat in a boiling water bath for 7 minutes. Cool, add 2 mL of boron trichloride–methanol solution (120 g in 1000 mL of methanol), cover with nitrogen, cap tightly, mix, and heat in a boiling water bath for 30 minutes. Cool to 40–50, add 1 mL of 2,2,4-trimethylpentane, cap, and mix on a vortex mixer or shake vigorously for at least 30 seconds. Immediately add 5 mL of saturated sodium chloride solution containing 1 volume of sodium chloride and 2 volumes of water. [Note—Shake from time to time. Before use, decant the solution from any undissolved substance, and filter if necessary. ] Cover with nitrogen, cap, and mix on a vortex mixer or shake thoroughly for at least 15 seconds. Allow the upper layer to become clear, and transfer to a separate tube. Shake the methanol layer once more with 1 mL of 2,2,4-trimethylpentane, and combine the 2,2,4-trimethylpentane extracts. Wash the combined extracts with two quantities, 1 mL each, of water, and dry over anhydrous sodium sulfate.
Test Solution 2 (for triglycerides)—
Transfer the equivalent amount of sample used to prepare Test Solution 1 to a 10-mL volumetric flask, and dissolve in and dilute with Internal Standard Solution to volume. Gentle heating (up to 60) may be applied to obtain a clear solution. Then proceed as directed in Test Solution 1, starting with “Transfer 2.0 mL”.
Test Solution 3 (for ethyl esters)—
In a 10-mL volumetric flask, dissolve the mass of sample to be examined, according to the table above, in the Internal Standard Solution, and dilute with the same solution to volume. Gentle heating (up to 60) may be applied to obtain a clear solution.
Test Solution 4 (for ethyl esters)—
Transfer the equivalent amount of sample used to prepare Test Solution 3 to a 10-mL volumetric flask, and dissolve in and dilute with Antioxidant Solution to volume.
Standard Solution 1—
Transfer 0.10 g each of USP Docosahexaenoic Acid Ethyl Esters RS and USP Eicosapentaenoic Acid Ethyl Esters RS, accurately weighed, to a 10-mL volumetric flask, and dissolve in and dilute with Internal Standard Solution to volume. Gentle heating (up to 60) may be applied to obtain a clear solution.
Standard Solution 2—
Transfer 2.0 mL of Standard Solution 1 to a glass tube, and evaporate the solvent with a gentle stream of nitrogen. Then proceed as directed for Test Solution 1 starting with, “Add 1.5 mL”.
System Suitability Solution 1—
Transfer 0.30 g of methyl palmitate, 0.30 g of methyl stearate, 0.30 g of methyl arachidate, and 0.30 g of methyl behenate, accurately weighed, to a 10-mL volumetric flask, and dissolve in and dilute with Antioxidant Solution to volume.
System Suitability Solution 2—
Transfer 55.0 mg of docosahexaenoic acid methyl ester and about 5.0 mg of tetracos-15-enoic acid (nervonic acid) methyl ester, accurately weighed, to a 10-mL volumetric flask, and dissolve in and dilute with Antioxidant Solution to volume.
Chromatographic System (see Chromatography 621)—
The gas chromatograph is equipped with a flame-ionization detector and a 0.25-mm × 25-m fused silica capillary column coated with a 0.2-µm film of G16. The temperature of the detector is maintained at 270 and that of the injection port at 250. The column temperature is initially set at 170 for 2 minutes, then increased at a rate of 3 per minute to 240, and is maintained at this temperature for 2.5 minutes. The carrier gas is helium with a split flow ratio of 1:200 and a flow rate of about 1 mL per minute. [Note—If splitless injection mode is used, solutions should be further diluted 1 in 200. ] Chromatograph System Suitability Solution 1 and System Suitability Solution 2, and record the peak responses as directed for Procedure: using System Suitability Solution 1, the area percent increases in the following order; methyl palmitate, methyl stearate, methyl arachidate, methyl behenate; the difference between the percent area of methyl palmitate and that of methyl behenate is less than 2.0 area percent units; using System Suitability Solution 2, the resolution, R, between docosahexaenoic acid methyl ester and tetracos-15-enoic acid methyl ester is not less than 1.2. [Note—In addition to the above system suitability requirements, the following is required for the analysis of triglycerides but not for ethyl esters. ] For triglycerides, chromatograph Standard Solution 1 and Standard Solution 2, and record the peak responses as directed for Procedure: the derivatization efficiency for the conversion of fatty acid ethyl ester to the fatty acid methyl ester is not less than 90.0% for each (DHA and EPA).
Procedure—
Separately inject duplicate equal volumes (about 1 µL) of Standard Solution 1, Standard Solution 2, Test Solution 1 (for triglycerides), Test Solution 2 (for triglycerides), Test Solution 3 (for ethyl esters), and Test Solution 4 (for ethyl esters) into the chromatograph, record the chromatograms, and measure the peak responses. Identify the retention time for the internal standard peak by comparing the chromatograms for Test Solution 1 and Test Solution 2 (for triglycerides) and by comparing the chromatograms for Test Solution 3 and Test Solution 4 (for ethyl esters). Calculate the percentage of EPA or DHA in the triglyceride taken by the formula:
100F(C/W)(RU/RS)
in which F is the factor to express the content of DHA (F = 0.921) and of EPA (F = 0.915) as free fatty acids; C is the concentration, in mg per mL, of either DHA or EPA in Standard Solution 2; W is the weight, in mg, of the sample taken to prepare Test Solution 1; RS is the ratio of peak responses of either EPA or DHA relative to the internal standard in the chromatogram of Standard Solution 2; and RU is the corrected peak response of either EPA or DHA relative to the internal standard in the chromatogram of Test Solution 1 calculated as follows:
1/(rU2/rT2 rU1/rT1)
in which rU2 is the peak response of any peak at the locus of the internal standard in the chromatogram of Test Solution 2; rU1 is the peak response of any peak at the locus of the internal standard in the chromatogram of Test Solution 1; rT1 is the peak response of EPA or DHA in the chromatogram of Test Solution 1; and rT2 is the peak response of EPA or DHA in the chromatogram of Test Solution 2. Calculate the percentage of EPA or DHA in the ethyl ester taken by the formula:
100F(C/W)(RU/RS)
in which F is the factor to express the content of DHA (F = 0.921) and of EPA (F = 0.915) as free fatty acids; C is the concentration, in mg per mL, of either DHA or EPA in Standard Solution 1; W is the weight, in mg, of the sample taken to prepare Test Solution 3; RS is the ratio of peak responses of either EPA or DHA relative to the internal standard in the chromatogram of Standard Solution 1; and RU is the corrected peak response of either EPA or DHA relative to the internal standard in the chromatogram of Test Solution 3, calculated as follows:
1/(rU2/rT2 rU1/rT1)
in which rU2 is the peak response of any peak at the locus of the internal standard in the chromatogram of Test Solution 3; rU1 is the peak response of any peak at the locus of the internal standard in the chromatogram of Test Solution 4; rT1 is the peak response of EPA or DHA in the chromatogram of Test Solution 4; and rT2 is the peak response of EPA or DHA in the chromatogram of Test Solution 3.
Content of Total Omega-3 Acids
Calculate the content of the total omega-3 acids by the formula:
EPA + DHA + ((An 3 × (EPA + DHA))/(AEPA + ADHA))
in which EPA is the content of EPA, in mg per g, obtained from the test for Content for EPA and DHA; DHA is the content of DHA, in mg per g, obtained from test for Content of EPA and DHA; An3 is the sum of the areas of the peaks corresponding to C18:3 n-3, C18:4 n-3, C20:4 n-3, C21:5 n-3, and C22:5 n-3 methyl esters in the chromatogram obtained with Test Solution 1 for triglycerides or the corresponding ethyl esters in the chromatogram obtained with Test Solution 4; AEPA is the area of the peak corresponding to the EPA methyl ester in the chromatogram obtained with Test Solution 1 for triglycerides or the peak corresponding to the EPA ethyl ester in the chromatogram obtained with Test Solution 4 for ethyl esters; and ADHA is the area of the peak corresponding to the DHA methyl ester in the chromatogram obtained with Test Solution 1 for triglycerides or the peak corresponding to the DHA ethyl ester in the chromatogram obtained with Test Solution 4.
WATER AND SEDIMENT IN FIXED OILS
Apparatus—
The preferred centrifuge has a diameter of swing (d = distance from tip to tip of whirling tubes) of 38–43 cm and is operated at a speed of about 1500 rpm. If a centrifuge of different dimensions is used, calculate the desired rate of revolution by the formula:
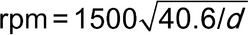
+'&img=../../images/v35300/g-1354-usp35.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
The centrifuge tubes are pear-shaped, and are shaped to accept closures. The total capacity of each tube is about 125 mL. The graduations are clear and distinct, reading upward from the bottom of the tube according to the scale shown in the accompanying table.
| Volume (mL) | Scale Division (mL) |
|---|---|
| 0–3 | 0.1 |
| 3–5 | 0.5 |
| 5–10 | 1.0 |
| 10–25 | 5.0 |
| 25–50 | 25.0 |
| 50–100 | 50.0 |
Procedure—
Place 50.0 mL of benzene in each of two centrifuge tubes, and to each tube add 50.0 mL of the oil, warmed if necessary to re-incorporate separated stearin, and mixed thoroughly at 25. Insert the stopper tightly into the tubes, and shake them vigorously until the contents are mixed thoroughly, then immerse the tubes in a water bath at 50 for 10 minutes. Centrifuge for 10 minutes. Read the combined volume of water and sediment at the bottom of each tube. Centrifuge repeatedly for 10-minute periods until the combined volume of water and sediment remains constant for three consecutive readings. The sum of the volumes of combined water and sediment in the two tubes represents the percentage, by volume, of water and sediment in the oil.
ANISIDINE VALUE
The Anisidine Value is defined as 100 times the optical density measured in a 1-cm cell of a solution containing 1 g of the substance to be examined in 100 mL of a mixture of solvents and reagents according to the method described below. [Note—Carry out the operations as rapidly as possible, avoiding exposure to actinic light. ]
Test Solution A—
Dissolve 0.500 g of the substance to be examined in isooctane, and dilute with the same solvent to 25.0 mL.
Test Solution B—
To 5.0 mL of Test Solution A add 1.0 mL of a 2.5 g per L solution of p-anisidine in glacial acetic acid, shake, and store protected from light.
Standard Solution—
To 5.0 mL of isooctane add 1.0 mL of a 2.5 g per L solution of p-anisidine in glacial acetic acid, shake, and store protected from light.
Procedure—
Measure the absorbance of Test Solution A at 350 nm using isooctane as the blank. Measure the absorbance of Test Solution B at 350 nm exactly 10 minutes after its preparation, using the Standard Solution as the compensation liquid. Calculate the Anisidine Value from the expression:
25(1.2AS AB)/m
in which AS is the absorbance of Test Solution B at 350 nm; AB is the absorbance of Test Solution A at 350 nm; and m is the weight, in g, of the substance to be examined in Test Solution A.
TOTAL OXIDATION VALUE (TOTOX)
Total Oxidation Value is defined by the formula:
2PV + AV
in which PV is the Peroxide Value, and AV is the Anisidine Value.
Change to read:
TRACE METALS
Apparatus
The apparatus typically consists of the following:
Digestion Flasks—
Use a polytetrafluoroethylene flask with a volume of about 120 mL, fitted with an airtight closure, a valve to adjust the pressure inside the container, and a polytetrafluoroethylene tube to allow the release of gas.
System—
Make the flask airtight, using the same torsional force for each of them.
Microwave Oven—
It has a magnetron frequency of 2450 MHz, with a selectable output from 0 to 630 ± 70 W in 1% increments, a programmable digital computer, a polytetrafluoroethylene-coated microwave cavity with a variable speed exhaust fan, a rotating turntable drive system, and exhaust tubing to vent fumes.
Atomic Absorption Spectrometer—
It is equipped with a hollow-cathode lamp as the source of radiation and a deuterium lamp as a background corrector; the system is fitted with the following:
- A graphite furnace as the atomization device for cadmium, copper, iron, lead, nickel, and zinc.
- An automated continuous-flow hydride vapor generation system for arsenic and mercury.
General Procedure
Caution—When using closed high-pressure digestion vessels and microwave laboratory equipment, the safety precautions and operating instructions given by the manufacturer must be followed.
[Note—If an alternative apparatus is used, adjustment of the instrument parameters may be necessary. ]
Cleaning—
Clean all the glassware and laboratory equipment with a 10 mg per mL solution of nitric acid before use.
Trace Metal–Free Nitric Acid—
Nitric acid meets the requirements with the maximum values for arsenic (As), cadmium (Cd), copper (Cu), iron (Fe), mercury (Hg), lead (Pb), nickel (Ni), and zinc (Zn) equal to 0.005, 0.005, 0.001, 0.02, 0.002, 0.001, 0.005, and 0.01 ppm, respectively.
Trace Metal–Free Hydrochloric Acid—
Hydrochloric acid meets the requirements with the maximum values for As, Cd, Cu, Fe, Hg, Pb, Ni, and Zn equal to 0.005, 0.003, 0.003, 0.05, 0.005, 0.001, 0.004, and 0.005 ppm, respectively.
Trace Metal–Free Sulfuric Acid—
Sulfuric acid meets the requirements with the maximum values for As, Cd, Cu, Fe, Hg, Pb, Ni, and Zn equal to 0.005, 0.002, 0.001, 0.05, 0.005, 0.001, 0.002, and 0.005 ppm, respectively.
Test Stock Solution—
In a digestion flask place about 0.5 g of fatty oil, accurately weighed, as indicated in each individual monograph. Add 6 mL of Trace Metal–Free Nitric Acid and 4 mL of Trace Metal–Free Hydrochloric Acid. Close the flask.
Blank Stock Solution—
Mix 6 mL of Trace Metal–Free Nitric Acid and 4 mL of Trace Metal–Free Hydrochloric Acid in a digestion flask.
Test Solution 1—
Place the digestion flask containing the Test Stock Solution in the microwave oven. Carry out the digestion in three steps according to the following program: 80% power for 15 minutes, 100% power for 5 minutes, and 80% power for 20 minutes.
At the end of the cycle allow the flask to cool. Add 4 mL of Trace Metal–Free Sulfuric Acid to the flask. Repeat the digestion program. After completing the digestion, allow the flask to cool to room temperature. Open the digestion flask, and transfer the clear, colorless solution obtained into a 50-mL volumetric flask. Rinse the digestion flask with 2 quantities, 15 mL each, of water, and collect the rinsings in the volumetric flask. Add 1.0 mL of a 10 mg per mL solution of magnesium nitrate and 1.0 mL of a 100 mg per mL solution of ammonium dihydrogen phosphate to the volumetric flask. Dilute with water to volume, and mix. This solution is Test Solution 1.
Blank Solution 1—
Place the digestion flask containing Blank Stock Solution in the microwave oven. Proceed as directed under Test Solution 1 beginning with “Carry out the digestion in three steps according to the following program”.
Direct Calibration—
[Note—Concentrations of the standard solutions will depend on the metal contents of the test substance. ] For routine measurements, three standard solutions, Blank Solution 1, and Test Solution 1 are prepared and examined.
Use Test Solution 1 and Blank Solution 1 as prepared above or as indicated in the monograph. Prepare not fewer than three standard solutions containing all the metal elements to be tested. The expected absorbance value in Test Solution 1 for each metal element should be within its corresponding calibrated absorbance range, preferably in the middle of the calibrated absorbance range. Any reagents used in the preparation of Test Solution 1 are added at the same concentration to the standard solutions.
Introduce each of the solutions into the instrument using the same number of replicates for each of the solutions to obtain a steady reading.
Prepare a calibration curve from the mean of the readings obtained with the standard solutions by plotting the means as a function of concentration. Determine the concentration of the element in Test Solution 1 from the curve obtained.
Standard Additions—
Add to at least four identical volumetric flasks equal volumes of Test Solution 1, as prepared above or as indicated in the monograph. Add to all but one of the flasks progressively larger volumes of a standard solution containing a known concentration of the test element to produce a series of solutions containing steadily increasing concentrations of that element known to give responses in the linear part of the curve. Dilute the contents of each flask with the solvent specified in the monograph to volume, and mix. The flask without an addition of standard solution is labeled as the test solution.
Introduce each of the solutions into the instrument, using the same number of replicates for each of the solutions, to obtain a steady reading.
Plot the absorbances of the standard solutions and the test solution versus the added quantity of test element. [Note—The test solution should be plotted as if it had a content of added test element equivalent to 0 mg or µg. ] Extrapolate the line joining the points on the graph until it meets the concentration axis. The distance between this point and the intersection of the axes represents the concentration of test element in the test solution.
Specific Tests
cadmium (cd), copper (cu), iron (fe), lead (pb), nickel (ni), and zinc (zn)
Standard Stock Solution—
Prepare a solution containing known concentrations of 5 µg per mL for each test element.
Standard Solutions—
In three identical 10-mL volumetric flasks, introduce 10, 20, and 40 µL of Standard Stock Solution, respectively. USP35 To each flask, add 5.0 mL of Test Solution 1, dilute with water to volume, and mix.
Test Solution 2—
In a 10-mL volumetric flask, USP35 add 5.0 mL of Test Solution 1, dilute with water to volume, and mix.
Blank Solution 2—
In a 10-mL volumetric flask, USP35 add 5.0 mL of Blank Solution 1, dilute with water to volume, and mix.
Procedure—
Measure the content of Cd, Cu, Fe, Pb, Ni, and Zn using a suitable graphite furnace atomic absorption spectrophotometer. Concomitantly determine the absorbances of Blank Solution 2, the Standard Solutions, and Test Solution 2 at least three times each. The absorbance value of Blank Solution 2 is subtracted from the value obtained using the Standard Solutions and Test Solution 2. Proceed as directed in the Standard Additions method in General Procedure above. See Table 1 for instrumental parameters that may be used.
Table 1
| Cd | Cu | Fe | Pb | Ni | Zn | ||
|---|---|---|---|---|---|---|---|
| Wavelength (nm) | 228.8 | 324.8 | 248.3 | 283.5 | 232 | 213.9 | |
| Slit (nm) | 0.5 | 0.5 | 0.2 | 0.5 | 0.2 | 0.5 | |
| Lamp current (mA) | 6 | 7 | 5 | 5 | 10 | 7 | |
| Ignition temperature ( |
800 | 800 | 800 | 800 | 800 | 800 | |
| Atomization temperature ( |
1800 | 2300 | 2300 | 2200 | 2500 | 2000 | |
| Background corrector | On | Off | Off | Off | Off | Off | |
| Nitrogen flow (L per minute) | 3 | 3 | 3 | 3 | 3 | 3 | |
arsenic and mercury
Measure the content of arsenic and mercury against their standard solutions of arsenic or mercury at a known concentration using the Direct Calibration method from the section General Procedure above, with an automated continuous-flow hydride vapor generation system.
For 1 ppm arsenic specification limit and 1 ppm mercury specification limit, prepare three working calibration solutions having known concentrations of 5 ng per mL, 10 ng per mL, and 20 ng per mL for each test element, respectively.
The absorbance value of the blank solution is automatically subtracted from the value obtained using the test solution.
Arsenic—
Blank Solution 3—
Add 1.0 mL of a 200 mg per mL solution of potassium iodide to 19.0 mL of Blank Solution 1 prepared above. Allow this solution to stand at room temperature for about 50 minutes or at 70 for about 4 minutes.
Test Solution 3—
Add 1.0 mL of a 200 mg per mL solution of potassium iodide to 19.0 mL of Test Solution 1 prepared above. Allow this solution to stand at room temperature for about 50 minutes or at 70 for about 4 minutes.
Acid Reagent 1:
Trace Metal–Free Hydrochloric Acid.
Reducing Reagent 1:
a 6 mg per mL solution of sodium tetrahydroborate in a 5 mg per mL solution of sodium hydroxide.
The instrumental parameters in Table 2 may be used.
Mercury—
Blank Solution 4—
Proceed as directed for Blank Solution 3.
Test Solution 4—
Proceed as directed for Test Solution 3.
Acid Reagent 2:
a 515 mg per mL solution of Trace Metal–Free Hydrochloric Acid.
Reducing Reagent 2:
a 10 mg per mL solution of stannous chloride in a 200 mg per mL solution of Trace Metal–Free Hydrochloric Acid.
The instrumental parameters in Table 2 may be used.
Table 2
| As | Hg | ||
|---|---|---|---|
| Wavelength (nm) | 193.7 | 253.7 | |
| Slit width (nm) | 0.2 | 0.5 | |
| Lamp current (mA) | 10 | 4 | |
| Acid reagent flow rate (mL per minute) |
1.0 | 1.0 | |
| Reducing reagent flow rate (mL per minute) |
1.0 | 1.0 | |
| Flow rate for the blank, standard, test solutions (mL per minute) |
7.0 | 7.0 | |
| Absorption cell | Quartz (heated) |
Quartz (unheated) |
|
| Background corrector | Off | Off | |
| Nitrogen flow rate (L per minute) | 0.1 | 0.1 | |
Change to read:
STEROL COMPOSITION
Separation of the Sterol Fraction
Reference Solution A—
Dissolve an accurately weighed quantity of cholesterol in chloroform to obtain a solution of 5% (w/v).
Developing Solvent System:
a mixture of toluene and acetone (95:5) or a mixture of hexane and ether (65:35).
Test Solution A—
Weigh accurately 5 g of the test substance into a 250-mL flask. Add 50 mL of alcoholic potassium hydroxide TS 2 (2 N alcoholic potassium hydroxide),USP35 and heat to gentle boiling with continuous vigorous stirring until saponification takes place (the solution becomes clear). Continue heating for a further 20 minutes, and add 50 mL of water from the top of the condenser. Cool the flask to approximately 30. Transfer the contents of the flask to a 500-mL separating funnel with several rinses of water, amounting in all to about 50 mL. Add approximately 80 mL of ether, shake vigorously for approximately 30 seconds, and allow to settle. [Note—Any emulsion can be destroyed by adding small quantities of ethyl or methyl alcohol by means of a spray. ] Separate the lower aqueous phase, and collect it into a second separating funnel. Perform two further extractions on the water–alcohol phase in the same way, using 60–70 mL of ether on each occasion. Pool the ether extracts into a single separating funnel, and wash with water, 50 mL at a time, until the wash water is no longer alkaline to phenolphthalein. Dry the ether phase with anhydrous sodium sulfate, and filter on anhydrous sodium sulfate into a previously weighed 250-mL flask, washing the funnel and filter with small quantities of ether. Distill the ether down to a few mL, and bring to dryness under a slight vacuum or in a stream of nitrogen. Complete the drying at 100 for approximately 15 minutes, and then weigh after cooling in a desiccator. Dissolve the unsaponifiables so obtained in chloroform to obtain a solution having a concentration of approximately 5%.
Test Solution B—
Treat 5 g of canola oil in the same way as prescribed for the test substance in Test Solution A, beginning with “Add 50 mL of alcoholic potassium hydroxide TS 2 (2 N alcoholic potassium hydroxide)”.USP35
Test Solution C—
Treat 5 g of sunflower oil in the same way as prescribed for the test substance in Test Solution A, beginning with “Add 50 mL of alcoholic potassium hydroxide TS 2 (2 N alcoholic potassium hydroxide)”.USP35
Procedure—
Immerse the thin-layer chromatographic plate (see Chromatography 621), 20-cm × 20-cm silica gel on polyester with a layer thickness of 200 µm and particle size of 5–17 µm,1USP35 completely in the 0.2 N alcoholic potassium hydroxide for 10 seconds, then allow to dry in a fume cupboard for 2 hours, and finally place at 100 for 1 hour. [Note—Remove from the validated heating device, and keep the plate in a desiccator until required for use. The plates must be used within 15 days. Thin-layer chromatographic plates without requiring the preconditioning are also commercially available. ] Use a separate plate for each test solution.
Place a mixture of toluene and acetone (95:5) or a mixture of hexane and ether (65:35) in the chamber to a depth of approximately 1 cm. Close the chamber with the appropriate cover, and leave for at least 30 minutes. Strips of filter paper dipping into the eluent may be placed on the internal surfaces of the chamber. [Note—The developing mixture should be replaced for every test to ensure reproducible elution conditions. ] Apply 0.3 mL of Test Solution A approximately 2 cm from the lower edge in a streak which is as thin and as uniform as possible. In line with the streak, place 2–3 µL of Reference Solution A at one end of the plate. Develop the chromatograms in an equilibrated chamber with the Developing Solvent System until the solvent front reaches approximately 1 cm from the upper edge of the plate. Remove the plate from the developing chamber, and evaporate the solvent under a current of hot air [Note—Avoid excessive heat. ] or by leaving the plate for a short while under a hood. Spray the plate with a 0.2% alcoholic solution of 2,7-dichlorofluorescein, and examine in UV light at 254 nm. [Note—The plates pretreated with UV indicator are also commercially available and used equivalently. ] In each of the plates, mark the limits of the sterol band identified through being aligned with the stain obtained from Reference Solution A along the edges of the fluorescence, and additionally include the area of the zones 2–3 mm above and below the visible zones corresponding to Reference Solution A. Remove the silica gel in the marked area into a filter funnel with a G3 porous septum.2USP35 Add 10 mL of hot chloroform, mix carefully with the metal spatula, filter under vacuum, and collect the filtrate in the conical flask attached to the filter funnel. Wash the residue in the funnel three times with ether, about 10 mL each time, and collect the filtrate in the same flask attached to the funnel. Evaporate the filtrate to a volume of 4–5 mL, transfer the residual solution to a previously weighed 10-mL test tube with a tapering bottom and a sealing stopper, and evaporate to dryness by mild heating in a gentle stream of nitrogen. Dissolve the residue in a few drops of acetone, and evaporate again to dryness. Place at 105 for approximately 10 minutes, allow to cool in a desiccator, and weigh.
Treat Test Solution B and Test Solution C the same way as directed for Test Solution A.
Determination of the Sterols
Test Solution D—
To the test tube containing the sterol fraction separated from the test substance by thin-layer chromatography, add a freshly prepared mixture of anhydrous pyridine, hexamethyldisilazane, and chlorotrimethylsilane (9:3:1) [Note—This reagent is also commercially available and used equivalently. ] in the ratio of 50 µL for every mg of sterols, avoiding any uptake of moisture. Insert the stopper into the test tube, and shake carefully until the sterols are completely dissolved. Allow it to stand for at least 15 minutes at ambient temperature, and centrifuge for a few minutes if necessary. Use the supernatant. [Note—The slight opalescence that may form is normal and does not cause an anomaly. However, the formation of a white floc or the appearance of a pink color is indicative of the presence of moisture or deterioration of the reagent. If these occur, the test must be repeated. ]
Reference Solution E—
To 9 parts of the sterols separated from canola oil by thin-layer chromatography, add 1 part of cholesterol. Treat the mixture in the same way as directed for Test Solution D.
Reference Solution F—
Treat the sterols separated from sunflower oil by thin-layer chromatography in the same way as directed for Test Solution D.
Chromatographic System (see Chromatography 621)—
The gas chromatograph is equipped with a flame-ionization detector and a glass or fused-silica capillary column of length 20–30 m, internal diameter 0.25–0.32 mm, entirely coated with a 0.10- to 0.30-µm layer of stationary phase G27 or G36. The injection port temperature is maintained at 280, the detector temperature is maintained at 290, and the column temperature is maintained at 260 ± 5. The carrier gas is either helium with a linear velocity of 20–35 cm per second or hydrogen with a linear velocity of 30–50 cm per second. A split ratio of 1:50 to 1:100 is used. Chromatograph Reference Solution E and Reference Solution F, and record the peak responses as directed for Procedure: the retention time should be 20 ± 5 minutes for  -sitosterol, and all the sterols present must be separated. The chromatogram obtained with Reference Solution E shows four principal peaks corresponding to cholesterol, brassicasterol, campesterol, and -sitosterol; and the chromatogram obtained with Reference Solution F shows four principal peaks corresponding to campesterol, stigmasterol, -sitosterol, and D7-stigmastenol. The retention times of the sterols with reference to -sitosterol are given in Table 3.
-sitosterol, and all the sterols present must be separated. The chromatogram obtained with Reference Solution E shows four principal peaks corresponding to cholesterol, brassicasterol, campesterol, and -sitosterol; and the chromatogram obtained with Reference Solution F shows four principal peaks corresponding to campesterol, stigmasterol, -sitosterol, and D7-stigmastenol. The retention times of the sterols with reference to -sitosterol are given in Table 3.
Table 3. Relative Retention Times of Sterols
for Two Different Columns
for Two Different Columns
| Identification | G36 Column | G27 Column |
|---|---|---|
| Cholesterol | 0.67 | 0.63 |
| Brassicasterol | 0.73 | 0.71 |
| 24-Methylene-cholesterol | 0.82 | 0.80 |
| Campesterol | 0.83 | 0.81 |
| Campestanol | 0.85 | 0.82 |
| Stigmasterol | 0.88 | 0.87 |
| D7-Campesterol | 0.93 | 0.92 |
| D5,23-Stigmastadienol | 0.95 | 0.95 |
| Clerosterol | 0.96 | 0.96 |
| 1.00 | 1.00 | |
| Sitostanol | 1.02 | 1.02 |
| D5-Avenasterol | 1.03 | 1.03 |
| D5,24-Stigmastadienol | 1.08 | 1.08 |
| D7-Stigmastenol | 1.12 | 1.12 |
| D7-Avenasterol | 1.16 | 1.16 |
Procedure—
Separately inject equal volumes (about 1 µL) of Test Solution D, Reference Solution E, and Reference Solution F into the chromatograph, record the chromatograms, and measure the peak areas for the sterols. Calculate the percentage of each individual sterol in the sterol fraction of the test substance taken by the formula:
100(A/S)
in which A is the area of the peak due to the sterol component to be determined, and S is the sum of the areas of the peaks due to the components indicated in Table 3.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Hong Wang, Ph.D.
Senior Scientific Liaison 1-301-816-8351 |
(GCCA2010) General Chapters - Chemical Analysis |
| Reference Standards | RS Technical Services 1-301-816-8129 rstech@usp.org |
USP35–NF30 Page 163
Pharmacopeial Forum: Volume No. 37(1)