



INTRODUCTION
This general information chapter describes good storage and distribution practices to ensure that drug products (medicines) reach the end user (practitioners and patient/consumers) with quality intact.
In the context of this chapter, the following definitions are used.
Definitions
Adulteration:
Continuous improvement:
Distribution:
Distribution Management System:
Documentation:
Drug products:
End user:
Environmental Management System:
Hazardous materials and/or dangerous goods:
International Conference on Harmonization (ICH) Guidance for Industry, Q10 Pharmaceutical Quality System; ICH Q9, Quality Risk Management; and, ICH Q1A R2, Stability Testing of New Drug Substances and Products:
Mean Kinetic Temperature (MKT):
Preventive actions:
Quality:
Quality Management System (QMS):
Risk Management System:
Written Agreement or Contract (commonly referred to as a Quality Agreement, Technical Agreement, Service Level Agreement, or other):
Storage Management System:
Supply chain:
Temperature stabilizer:
Transport vehicles:
SCOPE
Good storage and distribution practices apply to all organizations and individuals involved in any aspect of the storage and distribution of all drug products, including but not limited to the following:
- Manufacturers of drug products for human and veterinary use where manufacturing may involve operations at the application holder’s facilities (i.e., facilities that belong to the holder of an approved New Drug Application or Abbreviated New Drug Application) or at those of a contractor for the applicant holder
- Packaging operations by the manufacturer or a designated contractor for the applicant holder
- Repackaging operations in which the drug product may be owned by an organization other than the primary manufacturer
- Laboratory operations at the manufacturer’s or at the contractor’s site
- Physician and veterinary offices
- Pharmacies including but not limited to retail, compounding, specialty, mail order, hospital, and nursing home pharmacies
- Importers and exporters of Record
- Wholesale distributors; distribution companies involved in automobile, rail, sea, and air services
- Third-party logistics providers, freight forwarders, and consolidators
- Health care professional dispensing or administering the drug product to the end user
- Mail distributors including the U.S. Postal Service (USPS) and other shipping services including expedited shipping services
The information is intended to apply to all drug products regardless of environmental storage or distribution requirements.
It is recognized that conceivably there are special cases and many alternative means of fulfilling the intent of this chapter and that these means should be scientifically justified. Although this chapter is not intended to address the storage and distribution of active pharmaceutical ingredients (APIs), excipients, radioactive products, reagents, solvents, medical devices, medical gases, or clinical trial materials for which storage requirements may not yet be defined (e.g., Phase I clinical trial drug products), the general principles outlined here may be useful if applied selectively or comprehensively.
This general information chapter does not supersede or supplant any applicable national, federal, and/or state storage and distribution requirements, or USP monographs. General Chapter  659
659 Packaging and Storage Requirements contains definitions for storage conditions. This chapter is not intended to cover counterfeiting, falsified medicines, drug pedigrees, or other supply chain security, including chain of custody issues.
Packaging and Storage Requirements contains definitions for storage conditions. This chapter is not intended to cover counterfeiting, falsified medicines, drug pedigrees, or other supply chain security, including chain of custody issues.
BACKGROUND INFORMATION
Storage and distribution processes may involve a complex movement of product around the world, differences in documentation and handling requirements, and communication among various entities in the supply chain. The translation of best practices into good storage and distribution meets these challenges and sets forth a state of control.
The good storage and distribution practices described in this chapter should facilitate the movement of drug products throughout a supply chain that is controlled, measured, and analyzed for continuous improvements and should maintain the integrity of the drug product in its packaging during storage and distribution.
RESPONSIBILITIES
The holder of the drug product application, the drug product manufacturer (in the case of many OTCs, where there is no application) and the repackager bear primary responsibility and accountability including but not limited to the following:
- The decision for regulatory submissions, where applicable, relative to the contents of this chapter for the storage and distribution of drug products. If breaches occur in any of the QMS systems and cannot be justified or documented with scientific evidence, the appropriate entity should consider action with the product to ensure the public safety.
- Determining proper storage and handling practices
- Communicating storage and distribution practices through the supply chain
- Drug product stability profiles or the associated stability information from the holder, inclusive of distribution conditions and excursions that may be allowable should they occur. These stability profiles include the approved storage conditions for the shelf life of the drug product and, where appropriate, supporting data for the distribution conditions, if these differ from the storage conditions.
- Appropriate firms, such as an applicant holder, are to convey relevant environmental requirements (e.g., when appropriate, product-specific lifecycle stability data), when needed to support deviations or temperature excursions. If stability data cannot be reviewed or is not shared, an assessment may be needed to consider regulatory review or other appropriate actions (e.g., destruction of product or additional stability testing).
- Recalling the drug product if it is found to be adulterated in any part of the supply chain
However, all organizations along the supply chain bear responsibility for ensuring that they handle drug products within adequate storage and distribution parameters that will not affect the drug product identity, strength, quality, purity, or safety.
Each holder of drug product is responsible and accountable for the receipt from an entity and transfer out of the drug product to the next entity.
LABELING CONSIDERATIONS FOR DRUG PRODUCTS
The environmental requirements for drug product storage conditions should be indicated on the drug product primary container–closure system. If space on the immediate container is too small (e.g., an ampule) or is impractical for the container–closure system (e.g., blister package), this information can be placed on the most immediate container of appropriate size (e.g., carton). Environmental storage conditions and/or environmental warning statements should be evident, securely fixed, and indelible on the outermost container (generally the shipping container).
Products classified as hazardous materials and/or dangerous goods by the U.S. Department of Transportation or other relevant authorities or bodies should be labeled, stored, and handled in accordance with applicable federal/state/local regulations. Drug products classified as controlled substances by the U.S. Drug Enforcement Administration or by individual state requirements should be labeled and handled in accordance with applicable regulations.
Good practices and controls for labeling should provide the receiver with instructions for the correct handling of the drug product upon receipt. When a drug product’s storage conditions are not readily available, use the storage conditions described in USP’s General Notices and Requirements or the applicable USP monograph; or, contact the drug manufacturer for further information.
Product labels with expanded information beyond the single long-term storage temperature ensure ease of transport and use for shippers, distributors, healthcare professionals, and patients. Product labels should clearly define the storage temperature range, and broader distribution or in-use temperature ranges where allowable. Products labeled “Keep in a cold place” or “Do not freeze” are subject to interpretation and are discouraged if used without accompanying temperature ranges. USP storage definitions and temperature ranges are defined in General Notices and Requirements.
During international transport, the proper language(s) should be used to ensure that handlers understand the requirements set forth on drug product labeling. The use of symbols that are recognized by international organizations is advisable.
Drug products can be transported at temperatures outside of their labeled storage temperatures if stability data and relevant scientific justification demonstrate that product quality is maintained. The length of the stability studies and the storage conditions for a drug product should be sufficient to cover the shipment, distribution, and subsequent use of the drug product. The data gathered from ICH, Q1A R2, accelerated testing or from testing at an ICH intermediate condition may be used to evaluate the effect of short-term excursions outside of the label storage conditions that might occur during storage and/or distribution.
QUALITY MANAGEMENT SYSTEM
Good storage and distribution practices require that entities involved in the storage and/or distribution of drug products maintain a Quality Management System (QMS) that is based on standard quality concepts, includes good manufacturing practice (GMP) in compliance with the appropriate regulatory agency(s), and is complementary to the ICH quality guidances, including ICH Q10 Pharmaceutical Quality System and ICH Q9 Quality Risk Management. In the context of this chapter, the QMS includes the following management system programs: (1) Storage Management System, (2) Distribution Management System, (3) Environmental Management System, and (4) Risk Management System.
The storage and distribution QMS should, at minimum, cover the following elements: corrective and preventive actions (CAPA), change management, deviation/investigation management, and the management review process.
Written agreements (e.g., Quality Agreement, Technical Agreement, Service Level Agreements) should be in place between applicable organizations involved in the drug product supply chain. This means that the originating manufacturer may not be required to hold a Written Agreement with all parties in the supply chain. The use of written agreements ensures clarity and transparency, and delineates the responsibilities of each organization in the supply chain.
Good Documentation Practices
Good documentation practices should be practiced in the QMS. This documentation includes standard operating procedures and corporate policies and standards, as well as protocols and other written documents that delineate the elements of the QMS. The QMS programs should describe events and actions that must be documented as well as the proper verbiage to be used, the copies required, and any other items that will ensure adequate processing of the drug product and prevent delays. The documentation process should use a standard such as a quality manual or other practice and, should include routine assessment for review and update as needed.
Written procedures should ensure that drug products are held in accordance with their labeling instructions and associated regulatory requirements. Procedures should provide the written steps needed to complete a process and ensure consistency and standard outcomes. The following elements should be included: (1) how and when a product should be moved from one transport container/vehicle into another, (2) how products are handled when equipment malfunctions or when there are delays in distribution due to Customs hold, and (3) how to communicate with the necessary parties.
The QMS should require monitoring of processes to demonstrate that a state of control is being maintained, where the set of controls consistently provides assurance of continued process performance and product quality (ICH Q10).
If deviations occur, a nonconformance should be documented, and investigation should be performed and documented as appropriate. The investigative process should determine the root cause(s) of the deviation. For example, the following should be determined: whether the drug product experienced stress, damage, delays, or environmental lapses, or whether there were errors in documentation. The associated supply quality management staff should have final responsibility for approving or rejecting the investigation. The investigation process should be linked to the risk management program to ensure that proper mitigation occurs and preventive measures are put in place.
For example, a written investigation should be performed if the receiving and/or transferring processes result in a drug product being subjected to unacceptable temperature conditions or contamination (e.g., pests, microorganisms, or moisture). Any breach of standard operating procedures should be documented with a risk justification as needed. This information should be forwarded to the appropriate organization responsible for the drug product. The drug product should be quarantined, and final disposition should be based on good science with appropriate evidence to justify the decision(s).
Manufacturers should develop written procedures for recording the security process that confirms container–closure integrity for drug products that require special handling, such as security seals for controlled substances. Returned and salvaged goods records should address how the drug product is assessed through a written procedure. In addition, training on such procedures should be part of the QMS.
Records should be retained for purchases and sales of drug products and should show the date of purchase or supply; the name of the drug product and the amount; the name and address of the supplier or consignee; and the associated lot numbers. These records should allow for the traceability of a drug product in the supply chain.
All records and documents should be maintained in accordance with a traceable records-retention program and should be made available upon request to regulatory agencies. These documents should be approved, signed, and dated by the department responsible for the QMS.
Storage Management System
storage locations and processes
It is important that each entity define their appropriate storage locations to ensure that adequate controls are in place. These locations include buildings and facilities for drug product storage (e.g., warehouse, storage or hold area, the original manufacturer’s warehouses, contractor warehouses, wholesale distribution warehouses, mail order or retail pharmacy storage area, hospital or nursing home pharmacy storage areas; and border Customs storage areas).
In these locations, two basic processes can occur. First, receiving for storage is the act of bringing a drug product into a facility, while transferring refers to the moving of a drug product internally within a facility or into or out of a vehicle. Second, storing and holding refers to the act of maintaining temporary possession of a drug product in the supply chain process, during which no movement of the product will occur.
storage in buildings and facilities
Drug product storage areas are required to maintain the product temperature between the limits as defined on the product label. Buildings and facilities used for the warehousing, storage, and/or holding of drug products should be of adequate size for their intended use. These facilities should be adequate to prevent overcrowding. The building and facility should be designed to control environmental conditions where necessary and should be made of readily or easily cleanable materials. Sanitation and pest control procedures should be written, indicating frequency of cleaning and the materials and methods used. The pest-control program should ensure the prevention of contamination as well as the safe use of pesticides. Records of all cleaning and pest-control activities should be maintained.
Storage should be orderly and should provide for the segregation of approved, quarantined, rejected, returned, or recalled drug product. If computerized systems are used for the control of storage conditions, the software should be appropriately qualified for its intended purposes. Facilities should have controls that mitigate risks such as fire, water, or explosion. Certain drug products may cause these risks and should be stored accordingly. Storage areas, when not computerized, should be appropriately visually labeled.
Storage facilities themselves, unless thermostatically controlled, cannot be validated; however, they can be qualified via a mapping process. The generator back-up power supply should be qualified.
receiving and transferring drug products
Storage of a drug product includes not only the period during which the drug product is held in the manufacturer’s storage areas but also time spent at the receiving bay area. When drug products arrive at warehouse loading docks and other arrival areas, they should be transferred as quickly as possible to a designated storage or within a time period that is consistent with the risk and exposure of the product in the receiving area to a designated storage environment to ensure minimal time outside specified storage conditions as described in a written procedure.
Relative to the incoming receipt of drug product, it is recognized that the process of product reaction to ambient conditions begins immediately and may occur quickly (e.g., reach temperature equilibrium within minutes to a few hours depending on details such as the product mass, volume, and packaging density taking into account secondary and tertiary packaging)1. Time spent in a transport vehicle is considered to be part of the distribution process and is not a storage location.
Receiving docks should protect drug product deliveries from inclement weather during unloading. Any storage area, including loading and unloading docks for receipt and distribution of drug products, should be clean, cleanable, and free from pests. The incoming receiving area should limit access to authorized persons. Where appropriate, the delivery vehicle/container should be examined before unloading to ensure that adequate protection from contamination was maintained during transit. Deliveries should be examined at receipt in order to check that containers are not damaged and that the consignment corresponds to the order. The results of this examination should be documented.
Areas should be designated to provide an adequate space in which containers of drug products can be cleaned and opened for sampling. If sampling is performed in the receiving area, it should be done in a manner that prevents contamination and cross-contamination and ensures that environmental requirements for the drug product are not breached.
Adequate precautions should be taken to prevent theft and diversion of drug products. Drug products that have been identified as counterfeit should be quarantined to prevent further distribution. The appropriate regulatory agencies should be contacted according to established procedures.
Appropriate delivery records (e.g., as applicable, transport vehicle movement papers, receiving/delivery records, data logging records, temperature recorders and similar devices, bill of lading, house air waybill, master air waybill, etc.) should be reviewed by each receiving entity in the supply chain to determine if the product has been subjected to any transportation delays or other events that could have exposed the product to undesirable conditions. Each entity should ensure that their respective Service Level Agreement documents and supporting documents such as SOPs cover delivery and receiving responsibilities of the transactional parties.
Smoking, eating, and drinking should not be permitted in any storage/hold areas.
refrigerators and freezers
Refrigerators and freezers used to store drug products are required to maintain the product temperature between the limits as defined on the product label. Typically, a refrigeration unit specification would be set to 5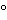 with an allowable range of ±3 to store products labeled 2–8. Freezer temperatures may vary and typically range from
with an allowable range of ±3 to store products labeled 2–8. Freezer temperatures may vary and typically range from  25 to 10. Some frozen drug products, however, require lower temperatures, e.g., dry ice or liquid nitrogen temperatures.
25 to 10. Some frozen drug products, however, require lower temperatures, e.g., dry ice or liquid nitrogen temperatures.
Regular operating procedures and maintenance protocols should be in place along with written contractual agreements for all maintenance and evaluation procedures including the following:
- Items should be stored in the units in a manner that allows adequate air flow to maintain the specified conditions.
- Units should be positioned in the facility so that they are not subjected to environmental extremes that could affect their performance. If this cannot be prevented, the mapping protocol should include a provision for testing during the anticipated environmental extremes.
- Large commercial units such as walk-in cold rooms are qualified via a temperature mapping study or other type of qualification process to determine the unit’s suitability for storing drug products. A suitable number of temperature-recording devices should be utilized to record temperatures and to provide temperature area maps. Thereafter, the units should be monitored as determined by the results of the mapping study. Refer to the Temperature Monitoring section under Environmental Management System.
- Units should utilize recording systems to log and track temperatures. Alarm systems should be an integral part of the monitoring system for both refrigerators and freezers. While automated systems monitor units continuously, manual checks should be performed as appropriate to the validation program. When automated systems are not available, manual systems may be used.
Distribution Management System
Distribution of drug products occurs within a facility or location such as a manufacturer, wholesaler, pharmacy dispensing area, retail site, clinic/hospital/nursing home pharmacy, and the physician’s practice. Distribution of drug products occurs as point-to-point movement within the supply chain between distribution facilities via semitrailer trucks, vans, emergency medical service vehicles, industry representatives’ automobiles, trains, aircraft, sea vessels, and mail delivery vehicles.
Communication within the supply chain should be coordinated to determine proper timing for drug products to be transported and received, taking into account holiday schedules, weekends, or other forms of interruption. When international distribution is required, alerts should be made in advance and proper language should be used to ensure understanding of the requirements set forth on drug product labeling.
packaging for the distribution and transportation processes
Pharmaceutical manufacturers should consider primary, secondary, and tertiary packaging that best protects the drug product during storage and distribution. Package performance testing should be documented as part of a manufacturer’s QMS. Several standard test procedures are available for evaluating package performance for factors such as shock, vibration, pressure, compression, and other transit events. Organizations with standard test methods include the following: the American Society for Testing and Materials (ASTM) Standard Practice for Performance Testing of Shipping Containers and Systems, and the International Safe Transit Association (ISTA) specifications for various types of transit modes such as less-than-truckload, small package, rail car, and air freight.
It is important to be aware that removal or modification of the original packaging may subject the product to unacceptable conditions.
The packaging (tertiary or thereafter) for the distribution of the drug product should be selected and tested to ensure that product quality is maintained and to protect the contents from the rigors of distribution including environmental or physical damage.
All drug products have storage requirements that may contain specific controls. The container used for transporting the drug product should be qualified on the basis of the labeled conditions of the product as well as anticipated environmental conditions. Consideration should be made for seasonal temperature differences, transportation between hemispheres, and the routes and modes of transport.
The type, size, location, and amount of the temperature stabilizers required to protect the product should be based on documented studies of specific distribution environments including domestic and international lanes, mode(s) of transport, duration, temperature, and other potential environmental exposures or sensitivities that may impact product quality. Transportation container materials such as warm/cold packs and materials used to control temperature conditions should be properly conditioned before use. Barrier protection may be important in helping to determine the position of materials such as gel packs in order to avoid direct contact with the drug product. It should be determined if studies are required to ensure that the dry ice and its vapors do not adversely affect the drug product, including the drug product labeling.
validation and thermal performance qualification for transport systems
Drug product transport systems should be continuously monitored by calibrated monitoring systems, (continuous verification), or shipping systems should be qualified and based on historical data relative to the process. However, it may be acceptable to use product stability data and supply chain risk assessment to justify shipping without either continuous monitoring or qualification of the shipping system.
Operational and performance shipping studies should on a generic level be part of a formal qualification protocol that may use controlled environments or actual field testing, depending on the projected transport channel. These studies should reflect actual load configurations, conditions, and expected environmental extremes. Testing should be performed on both active and passive thermal packaging systems.
Environmental Management System
While storage and distribution temperature ranges for drug products are labeled on the packaging, relative humidity effects occur over a much longer time frame. The primary container is designed and tested to protect the product from moisture; therefore, humidity monitoring should be considered when a product will be stored in an uncontrolled facility.
temperature monitoring
Environmental conditions are important parameters to consider in the storage and distribution of all drug products and may require monitoring depending on the requirements. When specific storage conditions are required and transportation qualification has not been performed, and in the absence of active or passive containers, environmental recorders or devices should be used to confirm that an acceptable range has been properly maintained during each stage in the supply chain.
Temperature is one of the most important conditions to control, and requirements for each drug product should be based on stability data. Temperatures should be tracked using a monitoring system, and the monitoring devices used should be included in a calibration and/or preventive maintenance program. Environmental monitoring devices should be calibrated for their range of operation. The monitoring devices used should provide an alert mechanism if the preset ranges are breached. The following practices and controls are examples of appropriate measures that should be put in place to ensure environmental control (see also Monitoring Devices—Time, Temperature, and Humidity 1118):
- Temperature-monitoring equipment, a monitoring device, a temperature data logger, or other such device that is suitable for its intended purpose should be used.
- An appropriate number of temperature monitors or some other form of recordation or proof of temperature control. Temperature monitor(s) should be used with every distribution process unless another process has been put in place to ensure specified temperature ranges.
- Electronic temperature monitors should be calibrated to National Institute of Standards and Technology (NIST) or other suitable standard.
- Chemical temperature indicators may be used as appropriate.
- Predetermined temperature ranges should be set for all applicable areas, as well as a plan of action in the event of an unacceptable excursion.
temperature mapping
The basis of any temperature mapping in a temperature controlled space (e.g., facility, vehicle, shipping containers, refrigerator, freezer) is the identification and documentation of a sound rationale used for a given mapping procedure. The temperature variability associated with mapped locations and the level of thermal risk to the product should be defined, unless another process has been put in place to ensure environmental control.
A temperature mapping study should be designed to assess temperature uniformity and stability over time and across a three-dimensional space. Completing a three-dimensional temperature profile should be achieved by measuring points at not less than three dimensional planes in each direction/axis—top-to-bottom, left-to-right, front-to-back, where product will be present.
When temperature mapping is necessary, it should begin with an inspection of the facility, equipment and/or vehicle and should be re-evaluated as appropriate. Environmental mapping also should be performed after any significant modification to the distribution system that could affect drug product temperature.
Facility temperature mapping:
The following factors, which may contribute to temperature variability, should be considered during the process of temperature mapping storage locations: (1) size of the space; (2) location of HVAC equipment, space heaters, and air conditioners; (3) sun-facing walls; (4) low ceilings or roofs; (5) geographic location of the area being mapped; (6) airflow inside the storage location; (7) temperature variability outside the storage location; (8) workflow variation and movement of equipment (weekday vs. weekend); (9) loading or storage patterns of product; (10) equipment capabilities (e.g., defrost mode, cycle mode); and (11) SOPs.
The recording of temperatures during the thermal mapping of a warehouse or cold room should be sufficient in time frame to capture workflow variation that may impact air flow and the resulting temperature fluctuation (i.e., a period of one week is recommended for data collection and should capture workflow cycles).
Equipment (container/trailer) temperature mapping:
To minimize risk of product exposure to damaging temperatures during transport, dedicated containers/vehicles cargo space should be mapped. When complete fleet mapping (i.e., wholesaler or distributor vehicles) is not realistic or appropriate, minimally at least one container/vehicle from the fleet must be mapped. Thereafter, the following conditions should be considered: (1) SOPs, including loading and unloading procedures; (2) route-specific operation of the temperature control equipment; (3) seasonal effects encountered on expected routes; (4) loading patterns; and (5) transport durations.
When nondedicated (i.e., mail carriers) transport containers/vehicles and equipment are used, they should be designed to minimize the risk of contamination of the product being handled. If environmental mapping of such vehicles is not performed, some other means of control should be in place to ensure that the drug product is adequately protected. Mapping by the shipper may not be necessary if the shipper uses a transport container that is properly insulated and has been previously qualified for the duration of the distribution process by the transport container manufacturer via a mapping study or if drug products are continuously monitored by calibrated monitoring systems (continuous verification).
The vehicle in which drug products are transported should be mapped to determine the appropriate placement of temperature-recording devices and to confirm that the load configuration is not restricting air flow. The following are recommended practices and controls for vehicles that receive and transfer drug products:
- Transport containers/vehicles and equipment used to store and transport drug products should be suitable for their intended function.
- Procedures should be established that describe how to operate, clean, and maintain transport containers/vehicles and equipment used in the storage and distribution of drug products.
- Transport containers/vehicles should be designed to prevent damage to the drug product, and pharmaceutical manufacturers should collaborate with their transporter to determine contingency response plans for how drug products are handled when equipment malfunction.
- When drug product must be moved from one transport container/vehicle into another, the proper load configuration should be followed.
- It should be understood how communication is made to the necessary entities when such transfer occurs.
- Subcontracted vehicles should be considered in contractual agreements and audits, and documentation should be maintained for their use.
Temperature mapping should account for maximum and minimum loads to capture temperature variability resulting from variations in temperature mass of the payload. Performance of equipment under extreme scenarios including door open, door closed, and simulated equipment failure should be taken into account.
Thermal mapping of vehicles should be representative of the fleet with the intention of capturing variability across the range of vehicles (type of vehicle including non-refrigerated equipment, use, heating and/or cooling system). A periodic requalification program should be documented.
Mapping for both facilities and transportation containers/vehicles should be done in a way that confirms their fitness for operation during periods of expected extreme weather (e.g., summer and winter). Facilities should be mapped under varying operating conditions—ideally during periods of greater variability, accounting for and capturing the result of any seasonal fluctuations of inventory movement, equipment movement, or workflow variation.
The temperature-mapping protocol and associated number of temperature data loggers used to map a three dimensional space should meet the intent of demonstrating three-dimensional uniformity and compliance with product requirements. For both facility and trailer/container temperature mapping, the ambient conditions should be recorded and correlations between ambient conditions and potential thermal risks inside the controlled space should be identified. Drug products should not be stored in areas where a thermal risk has been identified as a result of the temperature mapping. Areas identified as being unsuitable for storage should be clearly labeled as such to ensure that they are not used.
Temperature data loggers should be used for temperature mapping and PQ testing of facilities, equipment, and transportation containers used for storage or transportation of temperature-sensitive medicinal products. Temperature data loggers and any associated software applications should be appropriately validated. Certificates of calibration to an NIST or other international traceable standard should be available for individual monitoring devices.
excursions
The mapping process will help determine when excursions could occur and are useful when pharmaceutical manufacturers develop a plan for dealing with them. Alarms should be used to reveal environmental excursions during operations. Temperature excursions for brief periods outside of respective storage label conditions may be acceptable provided stability data and scientific/technical justification exists demonstrating that product quality is not affected (see Health Canada’s GUI 0069 entitled, Guidelines for Temperature Control of Drug Products During Storage and Transportation, 2011).
mean kinetic temperature (mkt) calculation
The MKT is the single calculated temperature at which the total amount of degradation over a particular period is equal to the sum of the individual degradations that would occur at various temperatures. MKT may be considered as an isothermal storage temperature that simulates the non-isothermal effects of storage temperature variation. It is not a simple arithmetic mean.
The temperatures used for calculating MKT can be conveniently collected using electronic devices that measure temperatures at frequent intervals (e.g., every 15 minutes). MKT can be calculated directly or the data can be downloaded to a computer for processing. Software to compute the MKT is available commercially.
For dispensing sites, such as pharmacies and hospitals, where the use of such instruments may not be feasible, devices such as high-low thermometers capable of indicating weekly high and low temperatures may be employed. The arithmetic mean of the weekly high and low temperatures is then used in the calculation of MKT. MKT is calculated by the following equation (derived from the Arrhenius equation):
+'&img=../../images/v38332/c1079-eq1-pf374.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
where Tk is the mean kinetic temperature; DH is the heat of activation, 83.144 kJ·mole–1 (unless more accurate information is available from experimental studies); R is the universal gas constant, 8.3144 × 10–3 kJ·mole–1·degree–1; T1 is the value for the temperature recorded during the first time period, e.g., the first week; T2 is the value for the temperature recorded during the second time period, e.g., second week; and Tn is the value for the temperature recorded during the nth time period, e.g., nth week, n being the total number of storage temperatures recorded during the observation period. [Note—All temperatures, T, are absolute temperatures in degrees Kelvin (K). ]
mkt during storage and distribution
The holding of a drug may occur as part of storage and distribution practices. Drug products in the distribution supply chain may be held at temperatures outside their labeled storage requirements as determined by an appropriate stability study. Drug products stored either in warehouse conditions or in transportation modes may experience excursions from their acceptable temperature ranges. Each product excursion must be evaluated to determine the final product effect. The means of evaluation must be scientifically sound with documented technical justification that the integrity of the drug product has not been affected. One method of analysis for drug product stored outside its respective label storage conditions is the use of an MKT calculation.
Because MKT expresses the cumulative thermal stress a drug product experiences, it is considered an acceptable practice for storage, and it follows that it should be considered for transit excursions in the process of distribution. The calculation must be justified for use with distribution excursions by confirming that the stability limiting characteristic of the product follows first order kinetics over the temperature range encountered. The ICH stability-testing guidelines define MKT as a “single” derived temperature, which, if maintained over a defined period, would afford the same thermal challenge to a pharmaceutical product as would have been experienced over a range of both higher and lower temperatures for an equivalent defined period.
The MKT analysis must be based on good science and should take into account the integrity of the product. The calculated MKT is not sensitive to the impact of excursions that may occur if the baseline is a long period of time such as a storage segment or the entire lifetime of the drug product. For shorter baseline periods of time, such as transport segments, an excursion can have a significant impact on the resulting MKT for that segment; however, this would not necessarily have a significant impact on product quality.
The MKT analysis may be used for storage conditions that have exceeded the acceptable parameters for a drug product, for a short period of time and is not intended to be a measure for long-term storage.
Knowing the MKT for an excursion is useful for evaluating the potential impact on product quality. However, it is also essential to know the upper and lower temperature limits of any excursion. If these extreme temperatures are outside available stability data, it may not be possible to predict the quality impact of the excursion with any confidence regardless of the MKT. Although higher temperatures are given greater weight in the calculation, the calculation of MKT for nonfrozen product that becomes frozen for any amount of time may not result in an acceptable temperature although the product may not be adulterated. At higher temperatures the kinetics of degradation may change or new degradation reactions may occur; at lower temperatures (near freezing) a phase change may occur that is known to have a negative impact on the quality of some drug products (e.g., some proteins and vaccines). For an example of a calculation, see Pharmaceutical Calculations in Prescription Compounding 1160.
Emergency Medical Service Vehicles, Automobiles, and Van Transportation
Road vehicles used to transport drug products (e.g., ambulances and other emergency response vehicles, vans, or automobiles, including those used by sales representatives to transport physicians’ samples) should be suitable for their purpose. Monitoring devices should be placed in different areas of the trunk or cabin where the drug product will be positioned during seasonal extremes (e.g., summer and winter). The monitor should be secured so that it is immobile, and there should be no ambiguity about its exact position within the payload so that the monitor is always placed in the same position. Monitoring devices used on or in packages or on containers may also be used. Suitable measures should be taken to maintain the drug product within the allowable limits of the labeled storage requirements. Storage of physician drug product samples by sales representatives is regulated under 21 CFR Part 203.34(b)(4).
Mail Order Pharmacy Distribution
The mailing party is accountable for the appropriate mailing process. Mail distributors including the U.S. Postal Service (USPS) and other shipping services including expedited shipping services are responsible to provide the service contracted.
In the event that the package cannot be delivered as scheduled, the package should be returned to the mailing pharmacy.
Risk Management System
Risk Management System strategies should ensure that each organization’s best interests are served by adhering to proper practices, controls, and procedures, including but not limited to the following: the nature of the drug products; distribution requirements on the readable container labeling; exposure to adverse environmental conditions; number of stages/receipts in the supply chain; manufacturer’s written instructions; contractors; and drugs at risk from freezing (vaccines, insulin, and biological products) or elevated temperatures (fatty-based suppositories, vaccines, insulin, and biological products).
Examples of risks include the following: (1) vibration that can cause aggregation of some drug products such as proteins and peptide-based drugs; (2) temperature excursions that may lead to phase changes (melting or freezing); (3) loss of container–closure integrity in transit that could cause glass fractures or loss of sterility in sterile drug product containers; and (4) ingress of water or oxygen that could lead to an increase in degradation products. Appropriate firms such as applicant holders are recommended to convey relevant environmental requirements when needed to support deviations or excursions. There may be alternate ways of determining acceptable environmental conditions and these should be documented and justified.
Pharmaceutical manufacturers should ensure that suppliers of drug product transportation are monitored. Auditing transportation firms should be carried out routinely to ensure adequate product handling. The manufacturer’s change control system should capture and evaluate changes in logistic factors such as warehouse or receiving areas and vehicle changes.
CONCLUSION
The practices and processes set forth in this general information chapter apply to storage and distribution as part of the life-cycle management of drug products. All involved should ensure the product to its point of use, creating a contiguous supply network that is collaborative and emphasizes preventive measures to protect drug product quality. The increase in global processes coupled with products requiring special environmental controls highlights the need for a strong QM program. QM should provide the foundation for maintaining the storage and distribution practices in a continual improvement program and part of an overall management system review by each entity, as appropriate, in the supply chain.
It is equally important to stay current and be ready to change as new solutions evolve. These new technologies should be considered in developing strategies for good distribution practices, controls, and procedures.
1
JP Emond, Study for Temperature Sensitive Product: Preliminary Testing, October 2009, University of Florida.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Desmond G. Hunt, Ph.D.
Senior Scientific Liaison (301) 816-8341 |
(GCPS2010) General Chapters - Packaging Storage and Distribution |
USP38–NF33 Page 1035
Pharmacopeial Forum: Volume No. 37(4)