



Introduction
Surface plasmon resonance (SPR) optical detection is a useful method for the label-free assays (procedures) that study biomolecular interactions. Commercially available SPR biosensors that incorporate these assays can collect real-time, information-rich data from binding events. These data can be used widely from basic research to drug discovery and development to manufacturing and quality control (QC). SPR can characterize binding events with samples ranging from proteins, nucleic acids, and small molecules to complex mixtures, lipid vesicles, viruses, bacteria, and eukaryotic cells. Typical quality and safety attributes addressed with SPR analysis include:
- Interaction specificity
- Interaction affinity
- Kinetic binding parameters
- Thermodynamic parameters
- Biologically active concentration of an analyte
This chapter provides an overview of the physics underlying SPR and common instrument configurations, as well as the range of molecules that can be studied and general considerations for experimental design as determined by the assay objective.
Overview
History
The physical principles of SPR were first explained in the early 1900s, starting with a description of the uneven distribution of light in a diffraction grating spectrum caused by the excitation of surface plasmon waves. A landmark series of experiments showed the optical excitation of surface plasmons under conditions of total internal reflection and fostered detailed studies of the application of SPR for chemical and biological sensing. Since then, SPR's potential for characterizing thin films and monitoring interactions at metal interfaces has been recognized, and significant research and development have yielded instruments that can quantitatively evaluate the binding interactions of small and large molecules.
Physics
SPR is an optical phenomenon that occurs when a thin conducting film is placed between two media that have different refractive indices. In many commercially available instruments, the two media are glass and the sample solution, and the conducting film is preferentially a gold layer applied to the glass, although other conducting metals such as silver have been used. The glass–metal component comprises a solid support that is often referred to as a sensor.
Light applied to the glass under conditions of total internal reflection produces an electromagnetic component that is called an evanescent wave. The evanescent wave penetrates the medium of lower refractive index (typically the sample solution) without losing net energy. The amplitude of the evanescent wave decays exponentially with distance from the surface, roughly one-half of the wavelength of the incident light (e.g., for a light source of 760 nm the evanescent wave penetrates approximately 300 nm).
For a specific combination of wavelength and angle of incident light, electron charge density waves called plasmons are excited in the gold film. As energy is absorbed via the evanescent wave, a decrease in the intensity of the reflected light at a specific angle (the SPR angle) is observed. Analysts can conduct an SPR experiment by fixing the wavelength and varying the angle of incident light.
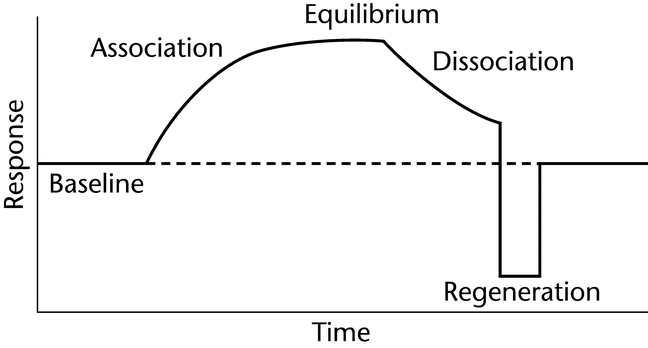
An increase in mass at the sensor surface caused by a binding interaction between two or more molecules causes a change in the local refractive index (RI) that gives rise to an SPR response, which is observed as a shift in the SPR angle. By monitoring the shift in the SPR angle as a function of time, an analyst can generate a sensorgram (Figure 1). The change in RI is very similar for different proteins, so the SPR measurement depends primarily on the mass change at the sensor surface and is relatively independent of the nature of the molecules being measured.
+'&img=../../images/v38332/m3070-f1-pf373.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 1. Representative sensorgram.
Instruments
The main components of commercially available SPR instruments are (1) a light source, typically a high-efficiency light-emitting diode, (2) an optical detector such as a diode-array or charge-coupled device camera, (3) a solid support containing the conducting film and some means for attaching molecules, (4) a sample delivery system, frequently a microfluidic device capable of delivering samples using single serial or parallel injections via single or multiple needles, and (5) a computer with appropriate software for instrument control, data collection, and analysis.
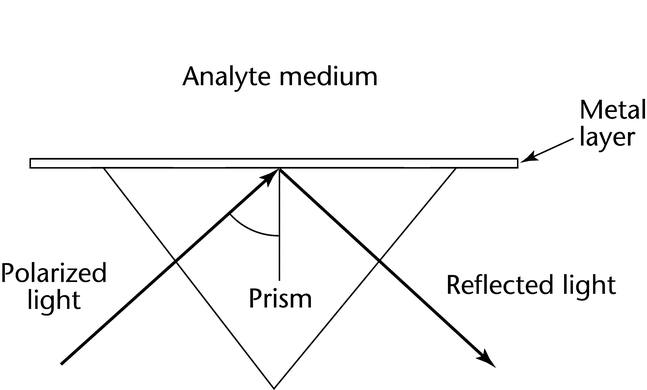
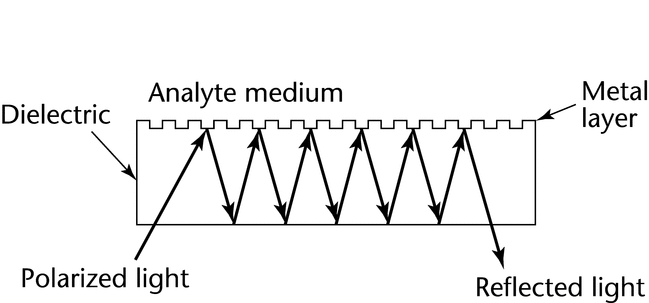
Prism-based and diffraction-grating instrument systems are commercially available. Most prism-based systems follow the Kretschmann configuration (Figure 2). The light is focused onto the sensor surface (away from the samples) via a prism with a refractive index matching that of the surface. In this configuration the incident light does not penetrate the sample solution, which permits SPR measurements for heterogeneous, turbid, or opaque samples. In systems that utilize a diffraction grating (Figure 3) the analyte solution is placed over a plastic surface on which a metal has been deposited. The plastic acts as an attenuated total internal reflection prism in which light reflected from the grating is reflected many times back to the grating surface. In this configuration light passes through the analyte sample solution, and thus turbid or opaque samples are not suitable for measurement. The diffraction grating does permit sampling of a larger surface area and is applicable for SPR measurements of arrays.
+'&img=../../images/v38332/m3070-f2-1susp35.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 2. Kretschmann SPR configuration.
+'&img=../../images/v38332/m3070-f3-1susp35.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 3. Diffraction grating SPR configuration.
The instruments are compatible with a wide range of biological samples and buffers as well as some organic solvents.
Biomolecular Interactions That Can Be Studied By Assays Using SPR
A diverse range of biological entities can be studied using SPR, including small molecules (<100 Da), proteins, nucleic acids, lipids, bacteria, viruses, and whole cells. Most published SPR research involves protein–protein interactions, of which antibody–antigen interactions represent a dominant subset. Improvements in instrument sensitivity and experimental protocols have helped analysts make studies of small molecules, lipids, and nucleic acids. Protein interactions with larger entities such as whole cells and some bacteria and viruses are limited by the exponential decay of the evanescent wave as described above. In practice these large molecules can be studied effectively, but the information obtained may be limited to qualitative or semiquantitative (e.g., relative ranking) data.
Assay Types
Several types of SPR assays are useful, including binding specificity, concentration analysis, kinetics and affinity analysis, and thermodynamics. Each assay type generates unique information that is helpful for profiling biomolecules.
SPR is also suitable for use in qualitative studies to confirm the specificity of interactions. Analysts can monitor a number of sequential binding events because each individual event yields a mass increase on the sensor chip surface, and all stages in the binding process are monitored. Examples include epitope mapping, antibody isotyping, and immunogenicity measurements.
Most chemical and spectroscopic methods used to quantify proteins (1) measure total protein content, (2) do not distinguish active from inactive molecules, and (3) cannot be used in conjunction with unpurified samples. Because SPR is a noninvasive method (no light penetrates the sample), it can measure small amounts of analyte molecules from complex matrices such as food products, serum or plasma, and cell extracts. Direct or indirect (inhibition or competitive) formats for measuring concentration are possible. SPR biosensors are uniquely suited for measurement of kinetic association and dissociation rate constants from real-time measurement of binding interactions. Affinity can be derived either from interactions that have reached equilibrium or from the ratio of the dissociation and association rate constants. The typical working range for affinity measurements is pM to high µM concentrations. Association rate constants that can be measured typically range from 103 to 107 M 1s1 and dissociation rate constants from 105 to 0.5 s1. By studying temperature dependence of rate and affinity constants, analysts can determine thermodynamic parameters for a binding interaction. Not only can the equilibrium values for changes in enthalpy (DH) and entropy (DS) associated with complex formation be determined, but transition state energetics can also be evaluated. Subsequent sections of this chapter address the specific details for these different assay types.
1s1 and dissociation rate constants from 105 to 0.5 s1. By studying temperature dependence of rate and affinity constants, analysts can determine thermodynamic parameters for a binding interaction. Not only can the equilibrium values for changes in enthalpy (DH) and entropy (DS) associated with complex formation be determined, but transition state energetics can also be evaluated. Subsequent sections of this chapter address the specific details for these different assay types.
The SPR Assay
The typical SPR assay involves five steps:
- Sample and buffer preparation
- Surface preparation
- Analyte binding
- Surface regeneration
- Data analysis and interpretation
Careful attention to experimental design leads to high-quality data and results. In SPR experiments, mass transport is essential for binding interactions to take place in instruments that use thin-layer flow-cell systems. Analyte molecules are transferred from the bulk solution to the binding surface via mass transport. When a limitation for binding occurs as a result of fast binding kinetics combined with high surface density, the binding interaction is considered mass-transport limited. In this case, the binding kinetics and complex formation are influenced by the availability of analyte molecules. The advantages and disadvantages of mass-transport–limited binding are discussed later in the application examples.
Sample and Buffer Preparation
Both purified and crude samples can be analyzed in a variety of matrices including serum, plasma, cell supernatants, and lysates. Crude samples containing particulates (e.g., cell debris or precipitates) may require clarification in order to help minimize unwanted binding. A short spin (30–60 s) in a benchtop centrifuge or filtration (0.22–1.0 µm) using low–protein-binding filters is recommended. The concentration range for evaluation depends on the experimental objective (yes/no binding, concentration, or kinetic/affinity analysis) as well as the binding affinity of the interacting molecules. In general, sample concentrations an order of magnitude below the equilibrium dissociation constant (KD), can be detected by SPR, but determination of an exact concentration is influenced by the analyte size (large vs. small molecules), binding specificity, and overall biological activity of the samples.
Most biological buffers and several organic solvents can be used in SPR experiments. The addition of salts and detergents to buffer solutions frequently can stabilize biomolecules. High-quality grade (e.g., molecular biology grade or higher) buffer components should be used. To simplify experiments, analysts should add only components that are absolutely required for biological activity or function. Buffers should be filtered and degassed before use.
Surface Preparation
Surface preparation involves the attachment of one of the binding partners to a solid support (surface). This process is frequently referred to as immobilization, and the resulting surface with the attached biomolecule is the sensor for the experiment. The choices of binding partner, solid support, and immobilization method are influenced by (1) the nature and demands of the application or experimental objective; (2) the availability of surfaces with different properties (e.g., charge density, hydrophobicity, or hydrophilicity); (3) the characteristics and supply of biomolecule to be used for immobilization; and, most importantly, that (4) biological activity be maintained and binding sites be available to interacting partners. Depending on the experimental objective, homogeneous or orientation-specific attachment of biomolecules also may be desired. The two main categories of immobilization methods are (1) direct immobilization, in which the molecule is covalently attached to the surface, and (2) indirect or capture immobilization, which takes advantage of tags or native groups on the protein or biomolecules (Table 1).
Table 1. Surface Preparation Techniques
| Chemistry | Immobilization Method | Biomolecules | Comments |
|---|---|---|---|
| Amine | Direct | Proteins, peptides | Amino terminus, Lys residues |
| Thiol—native | Direct | Proteins, peptides | Native Cys residue |
| Thiol—added | Direct | Proteins, peptides | Carboxyl groups derivatized |
| Aldehyde | Direct | Glycoproteins | Cis–diol required |
| Biotin capture | Indirect | Biotinylated peptides, nucleic acids, proteins | Stable, irreversible capture |
| Affinity tags | Indirect | Proteins, peptides | His, Glutathione S-transferase (GST), etc. |
| Protein A, Protein G | Indirect | Antibodies, IgG-tagged molecules | IgG species–dependent |
| Protein A, Protein G | Indirect | Biomolecules specific to the capturing antibody | Mono- or polyclonal antibodies may be suitable—testing recommended |
| Hydrophobic adsorption, membrane capture | Indirect | Lipids, membranes, membrane-associated proteins | Monolayer or bilayer attachment possible |
Direct Immobilization:
For direct immobilization, several chemistries are available for attaching proteins or other biomolecules to the surface. The properties of the surface determine the specific sequence of steps and length of time required to prepare the surface. Many commercially available surfaces have a biologically compatible layer (e.g., a hydrogel) that contains functional groups such as carboxyl that can be used for immobilization. To ensure binding specificity, the purity of the biomolecule that is attached to the surface should be 95% or greater and the required concentration should range from 1 to 1000 µg/mL. Direct immobilization chemistries frequently result in heterogeneous surfaces because of random orientation of biomolecules on the surface.
Immobilization via free primary amine groups such as lysine residues in proteins or the amino terminus of proteins or peptides is one of the most generally applicable covalent chemistries for attaching proteins to a surface. Carboxyl groups on the surface are converted to reactive esters using a mixture of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and N-hydroxysuccinimide (NHS) or sulfo-NHS (sNHS). The protein or biomolecule is applied in high concentrations (mg/mL) to maximize the efficiency of amine coupling. Finally, free esters are blocked with ethanolamine. The contact time with the surface, the protein concentration, or the EDC/NHS concentration can be varied to adjust the immobilization level.
When amine coupling interferes with the binding site, the biomolecule can be attached using alternative coupling chemistries or a high-affinity capture approach. For example, for biomolecules with free thiol groups (typically cysteine residues), a disulfide group is introduced by treating the surface with NHS and EDC to attach 2-(2-pyridinyldithio)ethaneamine (PDEA). Adding the biomolecule to the surface results in thiol–disulfide exchange, and excess PDEA groups are inactivated with cysteine–HCl. If the biomolecule lacks a free thiol group, a reactive disulfide (PDEA) can be linked to carboxyl groups. Subsequently the pyridyldisulfide groups can be attached to thiol groups on the surface that have been derivatized via injection of NHS and EDC, followed by cystamine, then reduction with dithioerythritol (DTE) or dithiothreitol (DTT). Attachment of maleimide groups to the surface makes possible an alternative form of immobilization via thiol groups in which a stable thioether bond is formed. Surfaces prepared using this method have the capacity to withstand basic pH (> 9.5) and reducing agents such as  -mercaptoethanol and dithiothreitol. Several heterobifunctional reagents are available commercially for introduction of reactive maleimido groups to the surface, including sulfo-MBS (m-maleimidobenzoyl-N-hydroxysulfosuccinimide ester), sulfo-SMCC (sulfosuccinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate), GMBS [N-(
-mercaptoethanol and dithiothreitol. Several heterobifunctional reagents are available commercially for introduction of reactive maleimido groups to the surface, including sulfo-MBS (m-maleimidobenzoyl-N-hydroxysulfosuccinimide ester), sulfo-SMCC (sulfosuccinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate), GMBS [N-( -maleimidobutyrloxy)sulfosuccinimide ester], EMCH [N-(
-maleimidobutyrloxy)sulfosuccinimide ester], EMCH [N-( -maleimidocaproic acid)-hydrazide] or BMPH [N-(-maleimidopropionic acid)-hydrazide]. For biomolecules containing either native aldhehyde groups or cis–diols, which may be converted into aldehydes by mild oxidation, surface attachment via a hydrazone bond is an option. Hydrazide groups on the sensor surface react with aldehyde groups on the biomolecule to form a stable bond. Immobilization via aldehyde groups is most useful for glycoconjugates, glycoproteins, and polysaccharides.
-maleimidocaproic acid)-hydrazide] or BMPH [N-(-maleimidopropionic acid)-hydrazide]. For biomolecules containing either native aldhehyde groups or cis–diols, which may be converted into aldehydes by mild oxidation, surface attachment via a hydrazone bond is an option. Hydrazide groups on the sensor surface react with aldehyde groups on the biomolecule to form a stable bond. Immobilization via aldehyde groups is most useful for glycoconjugates, glycoproteins, and polysaccharides.
Indirect (High-affinity Capture) Immobilization:
Indirect or high-affinity capture immobilization approaches use tags commonly used for protein purification. This technique exploits the high-affinity capture of the biomolecule by a capturing molecule that has been immobilized covalently using one of the techniques described above. The requirement for biomolecular purity is less stringent for indirect versus direct immobilization because the capturing step for the biomolecule can also provide purification. Indirect immobilization frequently yields a homogenous surface because all biomolecules are oriented similarly via the tag. The affinity between the biomolecule and its capturing agent should be sufficiently high to ensure little or no dissociation from the surface for the duration of an analysis cycle. Monoclonal antibodies are frequently used as capture molecules. For example anti-GST antibodies can be attached to the sensor chip surface via amine chemistry in order to capture GST-tagged molecules. Protein A, Protein G, and anti-IgG antibodies are useful capturing molecules for use with antibodies.
The high-affinity interaction between streptavidin or related molecules and biotin (KD 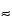 1015 M) makes it a useful system for the capture of biotinylated molecules (e.g., proteins, peptides, nucleic acids, membranes, and liposomes). Frequently, the biotin-binding protein is attached to the surface using primary amine groups. Because of the high affinity of the interaction, biotinylated molecules are considered permanently immobilized, and in contrast to most other capture approaches biotinylated molecules cannot be removed without damaging the surface. Histidine (His)-tagged recombinant proteins can be captured via nickel–NTA chemistry or covalently immobilized anti-His antibodies.
1015 M) makes it a useful system for the capture of biotinylated molecules (e.g., proteins, peptides, nucleic acids, membranes, and liposomes). Frequently, the biotin-binding protein is attached to the surface using primary amine groups. Because of the high affinity of the interaction, biotinylated molecules are considered permanently immobilized, and in contrast to most other capture approaches biotinylated molecules cannot be removed without damaging the surface. Histidine (His)-tagged recombinant proteins can be captured via nickel–NTA chemistry or covalently immobilized anti-His antibodies.
Lipids and membrane-associated proteins can be captured to the surface as either a lipid monolayer or bilayer. Lipids from micelles or liposomes adsorb to a hydrophobic surface, creating a lipid monolayer with the hydrophobic lipid tails oriented toward the solid support and the hydrophilic heads towards the aqueous sample. This approach provides a stable environment for proteins associated with a membrane surface or partially inserted into the membrane, but it is not ideal for transmembrane proteins because the resulting surface presents only half the membrane structure for binding interactions. Intact membrane structures (lipid bilayers) with associated or incorporated proteins can be captured by preparing liposomes with a specific antigenic component or with biotinylated lipids, allowing capture of the liposomes with immobilized antibody or streptavidin, respectively.
Additional Considerations:
Once the biomolecule has been attached to the sensor using either a direct or indirect immobilization approach, analysts should assess the baseline stability of the newly created surface. If the baseline is decreasing (downward drift), the most likely cause is the presence of unattached biomolecules, possibly because of self-association or aggregation. If the baseline is increasing (upward drift) refolding or re-orientation may be causing the change. In either case, the newly created surface should be conditioned before use by one or more of the following: (1) multiple injections of biologically compatible buffer; (2) washing the surface with buffer at a fast flow rate; (3) multiple injections of either high ionic strength (e.g., 1 M NaCl) or detergent (e.g., 20 mM CHAPS or 0.05% Polysorbate 20 (P20)) solutions; or (4) repeated analyte binding and regeneration injections. NOTE: recommendations (3) and (4) should be used only if the activity of the biomolecule in the presence of these reagents has been evaluated previously.
Large baseline drifts caused by low-affinity capture may be overcome by using EDC/NHS as a cross-linking step, but this may compromise biomolecule activity if active sites of the biomolecule are involved in the cross-linkages. The effect of cross-linking on biomolecule activity must be tested empirically for each biomolecule–analyte system. In general, cross-linking should be as brief as possible: 15 s is often sufficient to achieve acceptable baseline stability without compromising biomolecule activity.
How Much to Immobilize:
The amount of biomolecule to immobilize depends on the experimental objective. Equations 1 and 2 are useful for calculating the appropriate surface density:
Rmax = (MWA/MWL) × RL × Sm [Equation 1]
[Equation 1]
RL = Rmax × (1/Sm) × (MWL/MWA)[Equation 2]
| Rmax | = | = theoretical maximum binding response (assuming a surface that is 100% active and 100% bound with analyte) |
| RL | = | = response of the immobilized molecule |
| MWA | = | = molecular weight of the analyte |
| MWL | = | = molecular weight of the immobilized molecule |
| Sm | = | = molar binding stoichiometry |
For kinetic experiments, a low density of immobilized molecule is preferred in order to avoid steric hindrance, aggregation, and/or mass-transport–limited binding. Low density is defined as RL that limits Rmax to 5–50 response units. For other applications, e.g., concentration analysis where mass-transport–limited binding is desired, Rmax can be 100–200 times higher than for kinetic experiments provided that steric hindrance or aggregation are not induced. Specific recommendations for immobilization density are included in the application examples for this chapter.
Analyte Binding
Samples that will be evaluated for binding using SPR do not require the same purity as biomolecules intended for direct immobilization onto the surface. Because the light source does not penetrate the sample, turbid or opaque samples can be analyzed by SPR. Whenever practical, samples should be clarified according to the recommendations given under Sample and Buffer Preparation, and buffer additives should be minimized, including only the amount required for biological activity.
Differences between the refractive index of the bulk and sample buffers give rise to a response. The use of control surfaces and samples aids in demonstrating binding specificity for the molecules in SPR. For direct immobilization methods, suitable control surfaces can be (1) the sensor surface without any modification or biomolecule attached, (2) a surface that has been chemically treated in the same manner as the surface containing the biomolecule, or (3) a related but known nonbinding biomolecule. For surfaces prepared using indirect (capture) immobilization the capturing molecule in the absence of the tagged binding partner should be used as the control surface. The difference in response between the control and active surfaces gives an initial indication of the binding specificity. Concentration-dependent responses and inhibition of binding by incubating the sample with the biomolecule on the surface can further establish the binding specificity.
If nonspecific or unwanted binding is observed, analysts should determine the source. Frequently changes in pH or ionic strength of the buffers used in the experiment will reduce or eliminate the unwanted binding. Additional suggestions for reducing nonspecific binding are summarized in Table 2.
Table 2. Suggested Actions for Reducing Nonspecific Binding
| Category | Action |
|---|---|
| Experimental Design |
|
| Choice of Surface |
|
| Additions to Sample |
|
Equations 1 and 2 are also useful for assessing surface activity. The higher the binding response, the more active the surface is unless the observed binding response exceeds the calculated Rmax value. In this case, the molar binding stoichiometry is incorrect, the analyte molecule is aggregated, or the analyte is binding nonspecifically to the surface. Binding responses that are low (< 10% of Rmax) suggest that the analyte concentration selected for the experiment is too low or that the surface activity of the immobilized molecule is low. In the former case, increasing the analyte concentration should increase the binding response, and in the latter situation using a different immobilization method may be helpful.
Surface Regeneration
Surface regeneration refers to the process of removing bound analyte from the surface in order to reuse the surface for subsequent binding interactions. In some instances, complex dissociation is fast and bound analyte is simply washed away with buffer, so regeneration is not needed. Alternatively, the instrument configuration may allow multiple samples to be injected either sequentially or in parallel across several immobilized surfaces simultaneously, thereby limiting the need for regeneration. Inadequate surface regeneration may affect the reproducibility of an assay and negatively affect the overall quality of the resulting data. To identify the correct conditions, analysts should consider the nature of the specific interaction and the experimental objective. For example, a slight baseline drift will not affect the results when a simple yes/no answer is sought, but in concentration determination or kinetic studies, optimization of the regeneration step is critical.
Most biochemical interactions involve non-covalent bonds such as hydrogen, electrostatic, van der Waals, and hydrophobic bonds. Because the combination of physical forces responsible for binding and the regeneration conditions critical for not causing irreversible conformational changes are unknown for most interactions, the final conditions must be evaluated empirically.
The ideal condition for regeneration dissociates all the bound material without affecting the biological properties of the immobilized biomolecule. An incomplete regeneration or too stringent conditions may result in decreased analyte binding capacity in subsequent cycles because of either blocking of binding sites by nondissociated analyte or partial denaturation of the biomolecule. Regeneration buffers and solutions can be divided into different classes by the effect they have on the interaction. Any combination of buffers can be used.
The major classes of regeneration buffers are: acidic, basic, ionic/chaotropic, detergent, hydrophobic/nonpolar, and chelating (see Table 3 for examples of each class). Analysts should start with mild conditions, moving progressively to more harsh conditions. In many cases, especially when one is working with antibodies, change in pH is the most effective method of regenerating the surface. Contact time with the surface is important for efficient regeneration. When analysts use pH change, the contact times should be short, one-half to 2 min. When analysts use high ionic strength or chaotropes, longer contact times of 2–4 min are usually effective.
Table 3. Examples of Regeneration Solutions
| Acid | Base | Ionic/ Chaotropic |
Detergent | Hydrophobic/ Nonpolar |
Chelating |
|---|---|---|---|---|---|
| 1–100 mM HCl | 1–100 mM NaOH | 0.5–5 M NaCl | 0.02%–0.5% SDS | 25%–100% ethylene glycol | 10–20 mM EDTA or EGTA |
| 10–100 mM glycine, pH 1.3–3.0 | 10–100 mM glycine, pH 9.0–10.0 | 1–4 M MgCl2 | 40 mM octyleneglycol + 20 mM CHAPS | 5%–50% DMSO | 10–200 mM imidazole |
| 10–100 mM phosphoric acid | 1 M ethanolamine HCl, pH 9.0 or above | 1 M KSCN | 40 mM octylglucoside, 40 mM octylglucoside | 1%–10% acetonitrile | |
| 0.1% TFA | 100 mM sodium carbonate + 1 M NaCl, pH 9–11 | 2–6 M guanidine HCl | |||
| 100 mM Formic acid | 20–100 mM NaOH containing 0.5% surfactant P20 or 0.05% SDS |
The purpose of optimizing the regeneration conditions is to find the mildest possible regeneration solution that completely dissociates the complex. Analysts should maintain a constant level of activity over the binding–regeneration cycles even if the baseline changes a little. Repeated cycles of analyte binding followed by regeneration of the surface will provide insight into the overall performance of the surface. Ideally the surface performance should be evaluated for the same number of cycles that will be used during the SPR experiment.
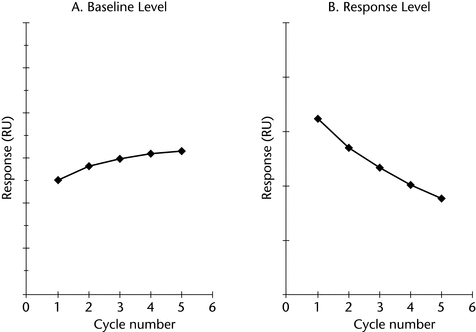
The surface must be monitored for signs of accumulation and also degradation of the immobilized ligand (Figure 4). This can be accomplished by monitoring both the baseline at the beginning of each injection cycle and binding signal (slope or bound response) of a quality control sample. Appropriately defined acceptance criteria for system suitability such as baseline drift and quality control performance help to monitor the integrity of the immobilized ligand on the surface.
+'&img=../../images/v38332/m3070-f4-1susp35.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 4. Evaluating surface performance (A) accumulation on surface and (B) degradation of immobilized ligand.
If the binding response is slowly decreasing, there are two possible explanations:
- If the baseline of the raw data sensorgram remains constant but the binding response still decreases, the regeneration conditions cause an irreversible change to the biomolecule that decreases the binding capacity of the surface, which in turn decreases the amount of analyte that can be bound on the surface. Analysts can decrease the strength of the regeneration solution slightly or can change to another regeneration solution of equal strength within the same class.
- If the baseline increases, the accumulation of analyte causes a binding-capacity decrease on the surface, which in turn decreases the amount of analyte that can be bound to the surface. Analysts can increase the strength of the regeneration solution slightly or can change to a regeneration solution of equal strength within the same class. There may be a difference between regeneration solutions in their ability to solubilize the analyte.
Once a suitable regeneration solution has been determined, it should be tested in a series of analyte binding and regeneration cycles. Because the binding activity of the surface typically decreases with time and/or use, analysts must empirically determine the binding response threshold and consequent number of cycles for surface use. Examples for determining binding threshold and number of cycles are presented below (see Applications 1–3).
In some cases the baseline will drop or rise and/or the binding capacity will decrease somewhat in the first few injections before it stabilizes. This is caused by either the dissociation of electrostatically bound biomolecules from the surface (depending on surface characteristics) or to binding to a high-affinity non-regenerable fraction of the surface. For this reason, each newly immobilized surface should be conditioned with repeated analyte binding and regeneration cycles before collection of quantitative data. Alternative immobilization methods, such as a different chemistry or indirect capture, should be evaluated when the immobilized biomolecule is difficult to regenerate.
Data Analysis and Interpretation
Analysis and interpretation of the data are specific to the experimental objective. Several data analysis programs exist to aid in the calculation of kinetics and affinity constants from SPR data. The validity and quality of the results are linked directly to experimental design. The fitting process is purely mathematical, without regard to the biological significance of the values obtained.
Data Analysis Algorithm:
Global analysis seeks a single set of kinetic rate constants for all of the analyte concentrations used in the experiment. Using a data-fitting algorithm such as Marquardt-Levenberg the data analysis software begins an iterative process starting with an initial approximation to find the best set of parameters that produces agreement between the experimental data (sensorgram) and the calculated fit to the data. The iterative process continues until the difference between the experimental and calculated (theoretical) curves is minimized as measured by the sum of the squared residuals.
Preparing the Data for Analysis:
Before conducting kinetic analysis, analysts should inspect the experimental data visually for anomalies or artifacts such as baseline disturbances or out-of-range data (often due to air bubbles) lasting for a predefined time period (e.g., 4–8 s). Outliers should be removed from the data set according to pre-established criteria. Nonessential data, such as capture or regeneration injections, should be removed from the sensorgram, and the data at each analyte concentration should be adjusted using the double-referencing procedure described below.
Before analysis the raw data should be processed in the following manner:
- Align the injection start to zero seconds for all concentrations and buffer injections for both the reference and active surfaces.
- Align the baseline to zero response for all sensorgrams.
- Subtract the reference surface sensorgram from the active surface sensorgram in order to create a corrected data set.
- Subtract the corrected buffer sensorgram from the sensorgrams at different concentrations in order to create a double-referenced data set.
The double-referencing procedure removes systematic errors (e.g., instrument noise) and low levels (less than 5% of total binding response) of nonspecific binding. It should not be used to correct for significant nonspecific binding events because this can lead to erroneous measurements.
When analyzing the data for kinetic information, analysts use the association (injection) and dissociation (buffer flow) phases for all of the concentrations in the series. For steady-state affinity analysis, the response at equilibrium Req (data plateau or no change in response vs. time) is measured for each sensorgram to create a binding isotherm with Req vs concentration. This isotherm is analyzed using the equations described below.
Kinetics and Steady-state Affinity Models:
The Langmuir kinetic model assumes a 1:1 interaction between the binding partners so that
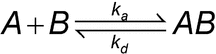
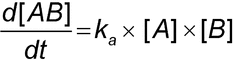
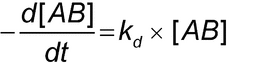
Association:dR/dt = ka × C × (Rmax R) kd × R
Dissociation:dR/dt = kd × R
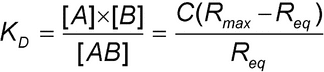
+'&img=../../images/v38332/m3070-eq3-pf373.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
The association and dissociation rate constants are defined below:
+'&img=../../images/v38332/m3070-eq4-pf373.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
+'&img=../../images/v38332/m3070-eq5-pf373.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Combining these two equations and defining [Bfree] = [Btot AB], the net rate expression is
d[AB]/dt = ka × [Afree] [Btot AB] kd × [AB]
which can be translated into terms from the SPR experiment as follows:
dR/dt = ka × C × (Rmax R) kd × R
where R is the binding response at any point along the sensorgram and C is the known analyte concentration. Using global analysis as described above, ka, kd, and Rmax are calculated from the experimental data using the rate equations shown below:
Association:
Dissociation:
Because the concentration of analyte is zero during dissociation, the rate equation for dissociation depends only on the response, R, and the dissociation rate constant, kd.
Application of the equilibrium condition where the complex formation (association) equals complex decay (dissociation)
ka × [A] × [B] = kd × [AB]
yields the following equation for the equilibrium dissociation constant
+'&img=../../images/v38332/m3070-eq10-pf373.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
where Req is the binding response at equilibrium that is measured in the experiment for a given analyte concentration and C, KD, and Rmax are calculated using global analysis.
Kinetics binding models can be used to describe non-1:1 interactions, e.g., bivalent interactions that occur if an antibody is used as the analyte, heterogeneity in binding partners, conformational change, or more complex interactions such as cooperative binding. SPR analysts are cautioned against using more complex models to assess data unless experimental design has been confirmed.
Assessing the Fit:
The quality and validity of the fit to the kinetic data can be assessed by (1) visual inspection of the agreement between the experimental and calculated curves, (2) the size and the shape of residual plots, (3) the biological relevance of the results, and (4) statistical parameters such as  2 (average of squared residuals), and standard error (SE), T-value or U (uniqueness) factor. The best parameter fit to the experimental data should be superimposed on the curve for each concentration in the experiment. The residual plot visualizes the difference between the calculated and experimental data. The shape of the residuals should be random without trending (waviness or curving up or down). The height of the residual plot should reflect the instrument noise. Further, 2 should be minimized for a good fit with values that depend on the instrument noise, number of data points, and overall binding response. The parameter values should be considered for biological and experimental relevance. For example, is the calculated ka value slow when the interaction is known to be fast, or is the calculated Rmax value higher than the value that was calculated using Equations 1 and 2. Parameter significance is evaluated based on standard error, T-values, and U factor. Parameters that are significant cannot be changed without affecting the quality of the fit. All of the criteria should be within acceptable limits.
2 (average of squared residuals), and standard error (SE), T-value or U (uniqueness) factor. The best parameter fit to the experimental data should be superimposed on the curve for each concentration in the experiment. The residual plot visualizes the difference between the calculated and experimental data. The shape of the residuals should be random without trending (waviness or curving up or down). The height of the residual plot should reflect the instrument noise. Further, 2 should be minimized for a good fit with values that depend on the instrument noise, number of data points, and overall binding response. The parameter values should be considered for biological and experimental relevance. For example, is the calculated ka value slow when the interaction is known to be fast, or is the calculated Rmax value higher than the value that was calculated using Equations 1 and 2. Parameter significance is evaluated based on standard error, T-values, and U factor. Parameters that are significant cannot be changed without affecting the quality of the fit. All of the criteria should be within acceptable limits.
A similar set of criteria can be used for assessing the fit to steady-state affinity data, but because there are fewer data points (6–12 total, depending on the number of concentrations used), the statistical parameters and residual plots are less predictive of fit quality. Visual inspection of the agreement between the experimental and calculated binding isotherms and the parameter relevance are good tools to use for assessing the fit. Additionally, according to the relationship between concentration, KD, and Rmax, when concentration equals the KD for the interaction R equals 50% of Rmax. Confirming that the analyzed data follow this relationship provides another way to check the validity of the calculated result. When it is practical to calculate the KD using both kinetic and steady-state analysis approaches, the KD values should agree within experimental error.
Addressing a Lack of Fit:
When the data do not fit or the parameter values do not make sense, often the problem can be resolved by a systematic approach that considers potential sources for deviations and tests each hypothesis. Items to consider include reagent purity, immobilization chemistry or surface density, analyte concentration errors, nonspecific binding, loss of ligand activity, or mass-transport–limited binding. Reviewing the raw (uncorrected) data helps determine the source of nonspecific binding or concentration errors. Table 4 lists potential sources for deviations from 1:1 binding and recommended actions.
Table 4. Common Sources for Deviation from 1:1 Binding Kinetics
| Source of Deviation | Recommended Action |
|---|---|
| Nonzero baseline before injection |
|
| Incorrect injection start and stop times or poorly defined injection start/stop |
|
| Concentration input errors |
|
| Bulk refractive index contribution too high |
|
| Mass-transport–limited binding |
|
| Nonspecific binding |
|
| Loss in binding partner activity |
|
| Multi-valent binding interaction |
|
Recommendations for data analysis will be introduced in the subsequent sections of this chapter.
Application 1—Immunogenicity Assessment:
SPR has emerged as a powerful technique for assessing immunogenicity of protein therapeutics. An advantage of this platform for detecting antibodies in serum (or plasma) samples is that it allows label-free detection based on mass accumulation in real time, which potentially allows detection of low-affinity antibodies of all classes and subclasses. This technology is useful both for screening assays (first-tier immunoassays that are used to detect the presence of antibodies capable of binding to a protein therapeutic) and also characterization assays. Characterization assays are useful for defining generated antibodies that bind to the protein and can include analysis of antibody concentration, isotype(s) represented, relative binding affinity, and binding specificity. A limitation is that SPR it is not appropriate for determination of the neutralizing capability of antibodies, which is best determined using cell-based biological assays. When designing and validating SPR assays for immunogenicity assessment, analysts should consider critical parameters including protein immobilization to ensure immunological reactivity, immobilized protein stability, and surface regeneration conditions.
Protein Immobilization:
The first step in the development of immunogenicity assessment assays is to identify the optimum mechanism for immobilization of the target protein. When considering the target density for immobilization, analysts often recommend that a high-density surface be used. The advantage of a high-density surface is that it maximizes the opportunity that anti-therapeutic antibodies will come in contact with an immobilized ligand. A high-density surface also provides excellent assay sensitivity. An important aspect of these assays is that the chemistry chosen for immobilization should provide random orientation rather than a site-directed orientation so that all potential epitopes on the therapeutic protein are available for binding by the anti-therapeutic antibodies. The effectiveness of immobilization is determined by evaluating the ability of positive control antibodies to bind to the immobilized protein. When evaluating the effectiveness of immobilization, analysts should test multiple antibodies with different epitope specificities. When panels of antibodies that cover a range of affinities and bind to different epitopes on the target protein are all capable of binding, this provides confidence that antibodies contained in clinical specimens also will be detected. If any of the positive control antibodies do not demonstrate binding, this suggests that the immobilization is not optimal and should be modified. Although SPR is demonstrably efficient at detecting low-affinity antibodies, analysts should confirm that the immobilization protocol chosen is effective for detection of low- and high-affinity antibodies.
Protein Stability Upon Immobilization:
The positive control antibody must be able to bind to the immobilized protein in order for an assay result to be acceptable. This confirmation of binding provides confidence that if antibodies against a protein are present in a sample, they will bind to the immobilized protein on the surface of the sensor. Because SPR relies on re-using the immobilized protein surface for multiple analyses, a regeneration protocol is required to effectively remove any bound material from the immobilized protein. This regeneration procedure is based on the ability to remove bound material without damaging or removing the immobilized protein. Several regeneration protocols can be used, and most often the regeneration solution is an acidic solution, commonly dilute HCl. The immobilized protein must remain intact and functional after repeated regeneration steps.
Because the immobilized protein will be used routinely for multiple analyses involving repeated cycles of serum samples, analysts should verify the stability of the immobilized protein after regeneration cycles. The stability of the immobilized protein can be monitored effectively by tracking the response units after regeneration and also after addition of positive control antibody. If there is a change in baseline or a decrease in the magnitude of binding by the positive control antibody, then the immobilized protein likely is no longer suitable for further analyses. The stability following regeneration should be established during assay development and should be confirmed during assay validation. In order to monitor the performance of the sensor during an assay, analysts should periodically test a positive control sample during an assay run. If the performance of the positive control samples indicates the immobilized protein has been compromised, analysts should re-analyze test samples obtained after the performance of the assay dropped below acceptable limits. Acceptance parameters for immobilization may vary by compound and should be established for each assay.
Availability of Epitopes After Immobilization:
Once the protein is immobilized, the availability of multiple epitopes should be confirmed. Ideally this is done by testing for binding of positive control antibodies with different epitope specificity. One method for testing epitope availability is to use a panel of monoclonal antibodies that are known to recognize different regions of the protein. If the protein has been randomly immobilized, all the different positive control antibodies should be able to bind. The reason for evaluating epitope availability is to prevent false-negative results when serum samples are evaluated. If the immobilization is not random, it would be possible to consistently immobilize the protein via a specific epitope, thus making that epitope unavailable for binding by an antibody. Another possibility is that chemical modification of the protein to facilitate immobilization altered the protein's conformation.
Surface Regeneration and Subsequent Protein Stability:
Using the previous guidelines, analysts should monitor the surface for signs of accumulation and degradation of the immobilized ligand and discontinue use when necessary. For example, when the binding capacity of a positive control antibody (diluted in test serum) drops below 80% of initial capacity the surface should not be used.
Assay Cut-point Determination:
When performing assays to determine if a serum sample contains antibodies against a protein, analysts sometimes observe a background level of binding. That background binding can vary depending on the nature of the immobilized protein and also the patient population being tested. In order to determine if a test sample contains antibodies, analysts compare binding to control samples that do not contain antibodies against the protein. A cut-point is established, and when a sample contains antibodies the binding is greater than that cut-point. Analysts determine the assay cut-point by analyzing a series of serum samples that do not contain antibodies against the immobilized protein and then performing statistical analysis to determine the level of binding consistent with a sample that does not contain antibodies. The cut-point should be established using the same conditions that will be used for sample analysis. Although different approaches are used for determining a cut-point, a common approach is to establish the mean from the binding of 50–100 serum samples from healthy volunteers and set the cut-point at 95% (equivalent to the mean plus 1.645 times the standard deviation for a normal distribution). Analysts should remove statistical outliers from the calculations because their inclusion can cause a high bias and raise the cut-point. This higher cut-point will result in identification of fewer samples with antibodies against the immobilized protein. The statistically evaluated cut-point is the response unit value that serum samples must exceed to be considered positive for the presence of antibodies against the therapeutic protein. An important feature of cut-point determination is that it may be different in different patient populations. For example, patients with inflammatory disease, may show a higher level of nonspecific reactivity compared to a normal population. This higher level of nonspecific binding would result in samples being identified as positive when they did not contain any antibodies specific for the protein. When this situation arises, predose serum samples can be used to establish a new patient population–specific mean and assay cut-point.
Analytical Procedure Development and Validation:
Once the stability of the immobilized protein is confirmed, a regeneration procedure has been defined, and the cut-point established, the antibody testing method can be developed and validated. The conditions used for analyzing samples should be identical to those used to establish the assay cut-point. An important parameter to consider is the optimal dilution of the serum sample. Increasing the dilution factor reduces nonspecific binding by serum proteins but also reduces overall sensitivity. Most antibody assessment procedures use between 5% and 50% serum. As the percentage of serum that is tested decreases, the percentage of the binding signal that is due to nonspecific interaction also decreases, and subsequently the percentage of the signal mediated by antibodies binding to the immobilized protein increases. Besides dilution, other means to reduce nonspecific interaction include adding surfactants, increasing salt concentration, adding BSA or HSA, or adding soluble sensor surface support material such as carboxymethyl dextran or alginate to the dilution and running buffer.
Other important variables to optimize include flow rate and sample volume. The combination of flow rate and sample volume defines the contact time, the length of time during which a given sample is in contact with the immobilized protein. The longer a sample is in contact with the immobilized protein, the greater the chances for antibody binding. The next important aspect to consider is verification that initial binding is a result of an antibody and not some other serum component. This can be accomplished by adding an anti-human immunoglobulin reagent and monitoring subsequent binding. If the initial binding observed was due to an anti-protein antibody, this reagent will bind to that antibody (the anti-protein antibody remains bound to the immobilized protein). When a therapeutic monoclonal antibody is the immobilized protein, the confirmatory reagent must be screened and verified not to bind directly to the immobilized therapeutic monoclonal antibody. One option here is the immobilization of the Fab' fragment rather than the intact therapeutic monoclonal antibody. The confirmatory reagent must be verified for specificity. Once all of the parameters are optimized, the assay can be validated. Validation parameters include those typically associated with immunogenicity assays (precision, specificity, sensitivity, and robustness) as well as parameters specific to SPR assays (protein immobilization, stability of immobilized surface, and number of regeneration cycles).
Interference by Serum Components:
Depending on the immobilized protein, serum components other than antibodies specifically directed against the immobilized protein possibly could bind to the immobilized surface. It is also possible that serum components that block the ability of antibodies to bind to the immobilized protein could be present. Both of these can be evaluated by testing the binding of serum samples from the target subject population that are known not to contain antibodies against the immobilized protein and then monitoring to determine if any binding does occur. If nonspecific binding is identified, steps can be taken to reduce or eliminate it. These steps can include pretreatment of samples to remove the nonspecific reactant, addition of surfactant, or alteration of salt concentration in sample buffers to reduce nonspecific binding.
Analysts should verify that serum samples do not contain agents that are capable of inhibiting antibody binding to the immobilized protein (these could include soluble forms of the immobilized protein or soluble receptors that could bind to the immobilized protein and block binding of the antibodies to the immobilized protein). Analysts can add the positive control antibody to target serum samples and can evaluate binding. If binding is inhibited by the target serum samples compared with binding to normal human serum samples, steps can be taken to remove the inhibiting agent. Failure to identify target serum interference can result in either false-positive or false-negative results.
Implementation of Multiplex Assays:
When a therapeutic protein is a second-generation product that has been modified from an original therapeutic protein (e.g., via pegylation or increased glycosylation), the presence of antibodies against both the original and the second-generation product should be evaluated simultaneously. This can be accomplished by immobilizing each protein on separate channels in the microfluidic device and allowing serum samples to bind in series or in parallel to both immobilized proteins. The rationale for testing for binding to both the original and the modified therapeutic protein is that antibodies generated against the modified protein could have binding specificity to the original protein as well. As part of the characterization of the immune response, analysts must understand the specificity of antibodies for both first- and second-generation products. When possible, binding to an endogenous counterpart might also be tested by immobilizing the endogenous protein on a separate flow cell or channel.
Characterization of Anti-Therapeutic Protein Antibodies:
Once antibodies against a therapeutic protein have been captured by binding to the immobilized therapeutic protein, those antibodies can be characterized. The important features of anti-therapeutic antibodies that can be studied include the relative binding affinity, the amount of antibodies present in the serum sample, the isotype(s) of antibodies present in the sample, and binding specificity.
By monitoring the rate at which the response units decrease after the conclusion of sample addition to the sensor, analysts can determine the relative affinity of the antibodies. A high rate of dissociation is characteristic of a low-affinity antibody, and a slow rate of dissociation suggests the presence of high-affinity antibodies. It is useful to compare the dissociation rates with both the positive control antibody (typically a high-affinity antibody preparation) as well as a panel of monoclonal antibodies of known binding affinities.
The relative active concentration of antibodies present in a sample can be estimated by comparing the binding signal with the signal produced from a dilution series of the positive control. Analysts can generate a standard curve from the standard and can compare the active concentration of antibodies in the sample with that standard curve. Because the positive control does not exactly mimic the mixture of antibodies contained in the sample—in fact, the positive control is often obtained from hyperimmunized animals such as rabbits—the concentration value obtained is relative to the standard. This value only approximates the actual concentration of human antibodies. Because the same positive control can be used throughout clinical development, analysts can compare the amount of antibodies obtained from different subjects using this strategy. Because the instrument's signal is proportional to the mass that is binding to the sensor, this type of analysis provides value. Analysts should consider that IgM antibodies have five times the mass of IgG antibodies. Another approach for determining the concentration of antibodies is described in the concentration analysis section of this chapter (see Application 2 below).
The isotype of captured antibodies can be readily determined by monitoring binding associated with sequential addition of isotyping reagents. For example, if IgM antibodies are present and have bound to the immobilized protein, the addition of an anti-human IgM reagent will produce an additional signal. Isotyping reagents can be found with specificity towards IgM, IgG, IgE, IgA, IgG1, IgG2, IgG3, and IgG4. Because of steric hindrance, analysts may be required to repeat isotyping analyses in different sequences to be certain the presence of previously bound isotyping reagents has not hindered subsequent analyses. For example, assume a sample contains both IgG1 and IgG4 antibodies against a protein and both species have bound to the immobilized protein. Because the anti-IgG1 isotyping reagent has bound to the IgG1 antibodies, the isotyping reagent bound to them may prevent subsequent additions of an anti IgG4 reagent from binding to the IgG4, and the presence of the IgG4 would be undiscovered. Analysts will conclude that only IgG1 antibodies are present, but if the order of isotyping reagent addition were reversed the IgG4 antibodies would be discovered. This example underscores the importance of careful interpretation of isotyping results. This is a problem for subsequent analysis only if there is observed binding by a previous cycle of isotype reagent addition. The specificity of isotyping reagents should be confirmed before use. Analysts should, for example, verify that an anti-human IgG reagent binds only to human IgG and does not cross-react with human IgM.
The region of the therapeutic protein recognized by the antibodies can sometimes be determined by immobilizing versions of the protein that have been truncated, have point mutations, or contain only a fragment of the protein. If the antibodies fail to bind to the changed version of the protein, it suggests that the epitope toward which the antibody is directed was influenced by the change. It should be kept in mind that point mutations and truncations not only influence the primary sequence of a protein, but can also influence the tertiary structure (i.e. folding, conformation) of a protein. Also, a subject is likely to generate a population of antibodies with different specificities for a variety of epitopes. Despite this concern, the strategy just described can prove useful for identifying the region on the protein where the antibodies are binding.
Application 2—Concentration Analysis:
SPR can be used to determine the concentration of biologics in defined buffer systems, e.g., eluates from purification columns, formulation buffers, and complex mixtures such as serum, fermentation broths, crude cell extracts, and cell suspensions. The concentration of an analyte is measured by its binding to the specific ligand or other molecules that can interact with any portion of the analyte. The analyte concentration is determined on a surface where the analyte-specific ligand or an analyte-specific capture reagent is immobilized. The binding rate or the mass of analyte bound is determined, and the analyte concentration is calculated using either a standard curve obtained from a concentration series of a purified and well characterized reference material or by a calibration-free analysis that is based on the relationship between the diffusion properties of the analyte and the absolute analyte concentration.
Immobilization of Ligand:
To determine concentration the ligand is immobilized covalently or non-covalently on the surface. Analysts select an appropriate coupling mechanism and chemistry to ensure the ligand's functional integrity. In order to provide conditions that favor partial or full independence of kinetic parameters, a high surface density of the ligand is desired.
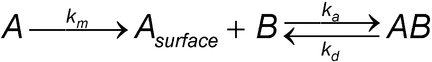
A high-density surface allows the analyte to bind to the ligand under conditions that limit mass transport. The interaction between the analyte and the ligand can be described by the following two-step process:
+'&img=../../images/v38332/m3070-eq11-pf373.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
where A is the concentration of analyte in the sample, Asurface is the concentration of analyte at the sensor surface, km is the mass transport coefficient, B is the immobilized ligand, AB is the analyte–ligand complex, and ka and kd are the rate constants for the reaction between A and B. The mass transport coefficient km depends on the flow rate, the dimensions of the flow cell, and the diffusion coefficient of the analyte. The analyte first must be transported from the bulk to the sensor surface to react with the immobilized ligand molecules. If this mass transport of the analyte is much faster than the association step between ligand and analyte, the overall observed binding will be driven by the kinetic rate constants of A and B, a prerequisite for accurately determining the kinetic parameters. If the mass transport of analyte is much slower than the association step, the binding will be limited by the mass transport process, and kinetic parameters for the specific interaction between A and B will be difficult to obtain. However, these conditions are desired for determining the active concentration of an analyte. A high density of ligand on the sensor surface and slower flow rates favor limited mass transport. Between these extremes, the overall binding is determined by contributions from both mass transport and interaction kinetics. It may not be possible to achieve limited conditions of mass transport for concentration analyses of interactions with relatively slow association rate constants (e.g., ka = 104 M1s1).
The ligand and the analyte reference material should be of sufficient purity with special attention to the presence of aggregated material. Aggregates of the analyte can interfere with the regeneration of the ligand surface because they can bind with multiple binding sites.
The reference material must be comparable (e.g., molecular weight and kinetic parameters) to the test samples. Under certain conditions the active concentration in unknown samples can be determined using a calibration-free procedure that is based on the relationship between the diffusion properties of the analyte and the absolute analyte concentration. These two methodologies are described separately below.
Concentration Determination with a Reference Standard Curve:
In typical concentration-determination assays the analyte concentration is calculated from a standard curve that is obtained with a reference material injected at select concentrations. Three different approaches can be used to measure concentration with a reference standard calibration curve:
Direct Binding Assay:
Determine the quantity of analyte bound after an arbitrarily fixed sample injection time. A sandwich method can be performed as an extension of the single-step direct binding approach in order to increase assay sensitivity.
Binding Rate Determination:
Determine the initial binding rate for a sample rather than the amount bound. Under conditions of mass-transport limited binding, the binding rate is directly proportional to analyte concentration, and is independent of binding kinetics. This allows one to measure the concentration of related molecules that might have different binding characteristics.
Inhibition or Competition Assays:
When the mechanism of action for an analyte is binding to a soluble ligand and thereby disrupting a ligand–receptor interaction, an inhibition assay can be used. In an inhibition assay, a receptor is attached to the sensor surface by a covalent linkage. The interaction between the analyte and the soluble ligand is indirectly measured by mixing a fixed concentration of ligand with varying concentrations of the analyte and injecting the ligand–analyte mixture across the immobilized receptor surface. Competitive methods in solution can also be used for large molecules and particles such as viruses, as well as for small analytes that give low direct responses. In parallel systems the assay can be designed so that the standard samples and the unknown sample are injected in parallel. This method can be useful for ligands that are difficult to regenerate.
Analysts can plot the signal (amount bound or rate of binding) of the reference material standards against concentrations and then can generate a standard curve using an appropriate mathematical model such as a linear or a logistic four-parameter curve fit. Samples can be injected at one or more dilutions. Fewer dilutions can be employed if a linear relationship between sample and reference standard has been demonstrated. Concentrations of unknown samples are either obtained by back-calculation from the standard curve or, if they are analyzed at the same target concentrations as those of the reference standard curve, by comparison of curve-fit parameters.
Parameters that can influence assay performance and results include but are not limited to flow rate, ligand density on the surface, sample purity, sample matrix, and reproducibility of surface regeneration. These parameters must be evaluated during assay qualification or validation. Interference with binding of analyte to the immobilized ligand can be minimized by salts, detergents, or sensor-surface support material. A commonly used sensor surface consists of carboxymethylated dextran, so the addition of dextran to the sample dilution buffer can minimize nonspecific interactions. Injections over a negative control surface can also be used to mathematically subtract the nonspecific binding data from the data obtained on the positive surface. A qualified or validated concentration determination SPR assay should include QC samples that can serve as measure to determine the accuracy of the standard curve that has been prepared to analyze samples with unknown analyte concentration. They can be conveniently prepared in larger batches, qualified for use with a Certificate of Analysis for the target concentration, and stored in small aliquots under appropriate storage conditions.
After each analyte injection the ligand surface is regenerated and all bound analyte is removed. This regeneration must be strong enough to remove all bound analyte, but the conditions also must leave the immobilized ligand intact so that injections can be compared to each other.
Concentration Determination Without Calibration:
Calibration-free concentration assays are based on the relationship between the diffusion properties of the analyte and the absolute analyte concentration. By measuring the initial binding rate analysts can derive the analyte concentration if specific properties of analyte and the analytical environment are known. This approach can be useful when no satisfactory reference standard is available.
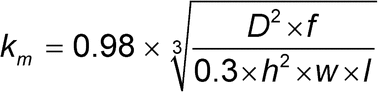
To determine the analyte concentration in a sample, analysts use the relationship between initial binding rate and analyte concentration. On a sensor surface with a high immobilization level, the initial binding rate (slope) can be described as a function of the molecular weight, the mass transport coefficient km, and the concentration of the analyte. Before a sample is analyzed analysts must determine the mass transport coefficient. It depends on the diffusion coefficient (D), flow rate, and flow cell dimensions and is described by the following formula:
+'&img=../../images/v38332/m3070-eq12-pf373.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
where D is the diffusion coefficient, f is the flow rate, and h, w, and l are the flow cell height, width, and length, respectively. Flow rate and flow cell dimensions typically are known for a given instrument, and the diffusion coefficient is determined by the size and shape of the molecule by the use of instrument-specific tools, literature references, or experiments, e.g., by analytical ultracentrifugation or light scattering.
In a typical experimental setup the evaluation requires two flow rates. By using measurements at two widely separated flow rates, analysts can assess the influence of flow rate on binding rate. The robustness of the assay is also improved by fitting the data obtained at two different flow rates, which give correspondingly two different values for km (because km depends on the flow rate), to a model with a global variable for analyte concentration (so that the model is constrained to find a single concentration value that best fits both curves simultaneously).
Calibration-free concentration analysis is suitable only for proteins with MW  5000 Da. It requires fast analyte–ligand association (ka > 5 × 104 M1s1) and it cannot handle mixtures of analytes with different diffusion properties. The dynamic range of the method is approximately 0.05–5 µg/mL.
5000 Da. It requires fast analyte–ligand association (ka > 5 × 104 M1s1) and it cannot handle mixtures of analytes with different diffusion properties. The dynamic range of the method is approximately 0.05–5 µg/mL.
Application 3—Kinetic and Affinity Analysis:
Because of its ability to detect binding interactions in real time, SPR provides valuable information about the kinetics of complex formation and dissociation. SPR instruments can be used to determine the association rate constant and dissociation rate constant for a particular binding interaction, and these values can be used subsequently to calculate the dissociation equilibrium constant (KD = kd/ka). Obtaining KD from a ratio of ka and kd is useful when the binding interaction does not reach equilibrium in a timeframe that is suitable for an SPR binding experiment. For binding interactions that reach equilibrium (rate of complex formation equals the rate of complex decay) in min (vs. h), KD can be determined directly from a steady-state binding response. The length of time required to reach equilibrium is influenced by the dissociation rate, so quickly dissociating complexes (e.g. kd = 102 s1) will reach equilibrium faster than those that dissociate slowly (e.g., kd = 105 s1). Software programs capable of simulating 1:1 binding kinetics are useful for predicting the length of time required to reach equilibrium. The typical working range for affinity measurements with commercially available SPR instruments is from 1012 M (pM) to 104 M (µM).
Proper experimental design is required to accurately measure ka, kd, and KD. Several questions must be considered when designing kinetic analysis or steady-state affinity experiments, including:
- Which binding partner should be immobilized?
- How will the analyst immobilize one of the binding partners?
- What type of reference surface should be used?
- How much binding partner should be immobilized?
- Does the binding partner maintain activity after immobilization?
- Is binding to the immobilized binding partner specific?
- What regeneration conditions, if necessary, should be used?
When selecting which binding partner to immobilize for most protein–protein interactions, analysts must consider several factors: (1) the purity and availability of the proteins, (2) the presence of a tag or functional group to aid in immobilization, (3) maintaining biological activity, and (4) the binding valency (e.g., monovalent vs. multivalent binding).
A reference surface is required for all detailed kinetic and affinity analysis experiments. If direct immobilization is used, then a reference surface is created using the same immobilization protocol, omitting the protein during the coupling step. Alternatively, a mutant form of the protein with a modified binding site can be used. The reference surface for high-affinity capture typically consists of either the capture molecule and no binding partner, or uses an unrelated molecule for a mock capture surface. For the specific case of antibody–antigen interactions, an unrelated monoclonal antibody often serves as the capture reagent on the reference surface.
After deciding on the immobilization approach, analysts must decide how much binding partner to immobilize. For kinetic analysis, the primary consideration is to minimize the surface density to avoid mass-transport–limited binding of the analyte molecule to the immobilized binding partner. Analysts also must consider the immobilization level when conducting steady-state affinity analysis because high immobilization levels can cause steric hindrance or can induce secondary effects such as nonspecific binding or aggregation.
Before performing a kinetic experiment or steady-state affinity analysis, analysts must assess the activity of the surface by injecting the analyte molecule at a single concentration. The concentration should be high enough that the equilibrium binding response provides a close approximation of the experimental maximum response (Rmax). This condition is typically met when the target molecule concentration is at least 10-fold higher than the KD of the binding interaction. For a protein–protein interaction having a KD value of 100 pM this means that the target molecule should be injected at a concentration of at least 1 nM. Using the Rmax equation (Equations 1 and 2), analysts can calculate the theoretical Rmax based on the amount of binding partner immobilized or captured. If the experimental Rmax exceeds the theoretical Rmax, then the analyte molecule is larger than expected or the analyte exists in a higher-order structure than expected (e.g., following aggregation or as a multimer). If the experimental Rmax is significantly lower (<50%) than the theoretical Rmax, this suggests that the immobilization procedure has compromised the binding site, and an alternative immobilization procedure should be investigated. An advantage of using high-affinity capture instead of direct coupling is that the surface activity typically remains close to 100%, provided that the specific activity of the immobilized binding partner is 100% before capture.
Injecting the target molecule across the reference surface also provides a quick assessment of the amount of nonspecific binding that exists on the sensor surface. Nonspecific binding that is electrostatic in nature can be eliminated or reduced by addition of salt (e.g., 0.5–1.0 M NaCl) to the sample diluent buffer and the running buffer or by using sensor surfaces with a low charge density. Nonspecific binding that arises from hydrophobic interactions can be minimized or eliminated by the addition of detergents such as 0.05% Polysorbate 20 or 10 mM CHAPS to the sample diluent buffer and the running buffer. Before using buffer additives, analysts should test whether the specific binding interaction or binding activity is affected. Nonspecific binding to a capture molecule can be resolved by switching to a different capture molecule. Reducing surface density also may eliminate nonspecific binding.
Many protein–protein interactions dissociate slowly (kd = 103 to 106 s1), with complex half-lives (t1/2) of more than 2 h. A regeneration step is important for these types of binding interactions.
In some protein–protein interactions, surface regeneration may not be possible because of a high-affinity binding interaction between the molecules. If this situation occurs, analysts can consider a titration kinetic experiment or the use of instruments that are capable of performing parallel analyte injections. In a kinetic titration, increasing concentrations of target molecule are injected consecutively across the immobilized surface, and the resulting data are analyzed using a titration kinetics model. In parallel instruments the analyte concentration series is injected in one step, thus eliminating the need for regeneration.
Surface regeneration typically is not performed for steady-state affinity experiments because the binding interactions in this type of experiment have kd values in the range of 102 to 0.5 s1, and therefore have t1/2 values of <2 min. The bound analyte dissociates from the surface as buffer flows over the surface, and regeneration is not required.
Analysts should have an accurate analyte concentration for kinetic analysis because the calculation of ka depends on the analyte concentration. Typically an absorbance reading at a wavelength of 280 nm (A280) is used for this purpose, but analysts should remember that the A280 value reflects the total bulk protein in solution and does not reflect the actual concentration of protein that is capable of binding (i.e., the active concentration).
Sample diluent injections should be included in replicate for kinetics and steady-state affinity experiments so that nonspecific responses due to instrumentation or sample diluent can be removed during the data-evaluation process.
The recommended analyte concentration range is 10-fold below and above the KD for the interaction. By keeping the surface density low (Rmax = 5–50 RU) and extending the association time, analysts should be able to collect data with enough curvature to accurately define ka and kd values. When the affinity of the interaction is high (KD = low nM to pM), higher analyte concentrations may be required to build a kinetic profile with sufficient curvature to complete the kinetic analysis. Increasing the analyte concentration should not be a substitute for changing other experimental design parameters (e.g., surface density and contact time). Although it is desirable to use analyte concentrations that approach Rmax, unusually high concentrations may induce aggregation of the analyte in solution or nonspecific binding to the surface.
Within an analyte concentration series, replicate samples should be used to assess surface activity. Additionally, each concentration series should be tested 3–5 times using different surfaces in order to establish confidence intervals for the resulting kinetics and affinity constants.
The amount of data that is collected during the kinetic or steady-state affinity experiment affects the accuracy of the results. For kinetic experiments involving binding interactions with slow kd values, analysts should collect enough dissociation data so that a measurable dissociation response (vs. instrument noise) is acquired. For example, a binding interaction between a therapeutic monoclonal antibody and its target molecule that has a kd of 10–5 s–1 will require the collection of at least 4 h of dissociation data. Rather than collecting this much dissociation data for all analyte concentrations, analysts can use a long dissociation time for the highest concentration of analyte that will be injected, and they can use a short dissociation time (2–5 min) for all other analyte concentrations. With parallel-injection instruments one can collect the long dissociation data for all of the concentrations. For data evaluation analysts should collect both long and short dissociation time data for sample diluent injections. For steady-state affinity experiments, the analyte injection time should be long enough to allow for a steady-state binding response to occur at all analyte concentrations that are tested. Dissociation data do not have to be collected for a steady-state affinity experiment because dissociation data are not used in the evaluation of this type of experiment, but the complete dissociation of analyte is required before beginning the next injection.
Use of SPR in a Regulated Environment
When SPR assays are used for lot release and stability testing, the assay must exist within a controlled setting so that decisions can be made about the use of product within the clinic or marketplace. SPR instrumentation, including software, should be 21 CFR Part 11 compliant and should be amenable to validation. These requirements are important because they help ensure the integrity of both data acquisition and data evaluation.
Besides using SPR instrumentation that meets regulatory requirements, analysts should establish system suitability criteria for an SPR assay. Including these criteria in an SPR assay ensures that the results obtained for the test sample are generated by an assay that is performing within its operating parameters. A discussion of assessing system suitability parameters is not within the scope of this chapter, but the reader is referred to USP general chapters Design and Analysis of Biological Assays  111
111 and Analysis of Biological Assays 1034 for more detailed discussions. Some examples of system suitability parameters for an SPR assay can include:
and Analysis of Biological Assays 1034 for more detailed discussions. Some examples of system suitability parameters for an SPR assay can include:
- ligand immobilization density
-
parameters from a curve fitting model (e.g. four parameter logistic curve fit)
— EC50 values for reference standard curve and positive QC curve— Effective asymptotes (response range) for reference standard curve and positive QC curve— R2 values for reference standard curve and positive QC curve— parallelism between reference standard curve and positive QC curve
- relative bioactivity for positive QC (EC50 ratio of reference standard to positive control)
- calculated concentration for positive QC (single-point positive QC measurement)
- binding response for negative QC (nonspecific analyte or diluent)
Multiple lots of ligand, analyte, coupling reagents, and sensor surfaces should be used to establish assay system suitability criteria that reflect normal assay conditions. Assay results for reference standard and QC samples should be tracked over time. Regular trending analyses should be done on the data to show whether the SPR assay remains in control over its required lifecycle. If trending in the data is observed, remedies can be performed proactively, preventing assay failure.
For SPR assays that are used in lot-release testing, it is also important to establish sample acceptance criteria. These criteria are used to accept or reject sample data and can include:
- relative bioactivity for test sample
- coefficient of variation (CV) for sample replicates
- parallelism between reference standard curve and positive QC curve
Another point to consider for SPR assays in regulated environments is the identification of noncritical and critical reagents. Noncritical reagents typically include coupling buffer, regeneration buffer, and continuous flow (running) buffer. Critical reagents typically include the ligand and analyte for direct binding assays, and ligand, analyte, and competitor molecule in inhibition binding assays. Critical reagents should be qualified/requalified on a routine basis to ensure that they are suitable for use in the SPR assay. Best practices for the characterization of critical reagents are not within the scope of this chapter, so the reader is referred to current regulatory documents for such discussion. Analysts should use current regulatory documents such as ICH Guideline Q2(R1) Validation of Analytical Procedures and USP–NF General Chapter Validation of Compendial Procedures 1225 when they validate SPR assays in a regulated laboratory.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Maura C Kibbey, Ph.D.
Senior Scientific Liaison, Biologics & Biotechnology (301) 230-6309 |
(GCBA2010) General Chapters - Biological Analysis |
USP38–NF33 Page 1146
Pharmacopeial Forum: Volume No. 37(3)