



PRINCIPLES OF NMR
Nuclear magnetic resonance (NMR) spectroscopy is an analytical technique based on the magnetic properties of certain atomic nuclei. NMR is similar to other types of spectroscopy in that absorption of electromagnetic energy at characteristic frequencies provides analytical information. NMR differs from other types of spectroscopy because the discrete energy levels between which the transitions take place are created by placing the nuclei in a magnetic field of strength H0. Although the initial field strength of the applied field is H0, when the sample is inserted into the magnet, the field strength throughout the sample becomes B0, defined as follows:
B0 = µSH0 [1]
[1]
in which µS is the magnetic susceptibility of the sample.
Atomic nuclei behave as if they were spinning on the nuclear axis. The angular momentum,  0, of the nucleus is characterized by a spin quantum number (I). The maximum observable component of the angular vector, , is
0, of the nucleus is characterized by a spin quantum number (I). The maximum observable component of the angular vector, , is
in which h is Planck’s constant and h is modified Planck’s constant.
Table 1 shows the values of I as a function of the mass number and the atomic number.
Table 1. Nuclear Spin Values as a Function of Mass and Atomic Numbers
| Mass Number |
Atomic Number |
Nuclear Spin (I) |
|---|---|---|
| Odd | Even or odd | 1/2, 3/2, 5/2 ... |
| Even | Even | 0 |
| Even | Odd | 1, 2, 3 ... |
The angular momentum creates a magnetic moment, µ, which is parallel to and directly proportional to .
µ =  = Ih [3]
= Ih [3]
where is the magnetogyric ratio and is a constant for all nuclei of a given isotope, regardless of their position in a molecule.
Nuclei that have a spin quantum number I  0, when placed in an external uniform static magnetic field, align with respect to the field in (2I + 1) possible orientations. Thus, for nuclei with I = 1/2, which includes most isotopes of analytical significance (Table 2), there are two possible orientations, corresponding to two different energy states. The energies of these two states are ± µB0, and their separation is
0, when placed in an external uniform static magnetic field, align with respect to the field in (2I + 1) possible orientations. Thus, for nuclei with I = 1/2, which includes most isotopes of analytical significance (Table 2), there are two possible orientations, corresponding to two different energy states. The energies of these two states are ± µB0, and their separation is
E = µB0  ( µB0) = 2µB0[4]
( µB0) = 2µB0[4]
with more nuclei populating the lower energy state (µB0) than the higher energy state (+µB0). The populations are in accordance with the Boltzmann distribution:
N+/N = exp(E/kT)[5]
where N+ and N are the populations of the high and low energy states, respectively; k is the Boltzmann constant; and T is the temperature in K.
A nuclear resonance is the transition between these states, and upward as well as downward transitions are possible. In a static magnetic field the nuclear magnetic axis precesses (Larmor precession) about the B0 axis. The precessional angular velocity is often referred to as the Larmor frequency, 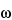 0, and is related to B0:
0, and is related to B0:
+'&img=../../images/v38332/c1761-eq6-usp36.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
If energy from an oscillating radio-frequency (rf) field is introduced, then resonance is achieved when the rf frequency is the same as the precessional angular velocity. Although the probability of an upward transition is equal to that of a downward transition, more upward transitions take place than downward transitions because N is greater than N+. Hence, an overall absorption of energy takes place. As shown in Table 2, the resonance frequency of a nucleus increases in direct proportion with the increase of the magnetic field strength.
Table 2. Properties of Some Nuclei Amenable to NMR Study
| Resonance Frequency (MHz) at |
||||||
|---|---|---|---|---|---|---|
| Nucleus | I | Natural Abundance (%) |
Sensitivity | 1.4093 Ta | 7.0463 T | 11.7440 T |
| 1H | 1/2 | 99.98 | 1.00 | 60.000 | 300.000 | 500.000 |
| 13C | 1/2 | 1.108 | 0.0159 | 15.087 | 75.432 | 125.721 |
| 19F | 1/2 | 100 | 0.83 | 56.446 | 282.231 | 470.385 |
| 31P | 1/2 | 100 | 0.0663 | 24.289 | 121.442 | 202.404 |
| 11B | (3/2) | 80.42 | 0.17 | 19.250 | 96.251 | 160.419 |
|
a
T = tesla: 1.4093 T = 14.093 kilogauss.
|
||||||
NMR is a technique of high specificity but relatively low sensitivity. The basic reason for the low sensitivity is the comparatively small difference in energy between the upper and lower energy states (0.08 Joules at 1.5 to 2.0 T field strength), which results in a population difference between the two levels of only a few parts per million. Another important aspect of the NMR phenomenon, with negative effects on sensitivity, is the long lifetime of most nuclei in the excited state. Long lifetimes affect the design of the NMR analytical test, especially in repetitive pulsed experiments. Simultaneous acquisition of the entire range of resonant frequencies instead of frequency-swept spectra can give increased sensitivity per unit time.
All characteristics of the signal–chemical shift, multiplicity, linewidth, coupling constants, relative intensity, and relaxation time contribute analytical information. The analytical usefulness of NMR arises from the observation that the same types of nuclei, when located in different molecular environments, exhibit different resonance frequencies. The reason for this difference is that the effective field associated with a particular nucleus is a composite of the external field provided by the instrument and the field generated by the circulation of the surrounding electrons. The latter field is generally opposed to the external field and lowers the overall field strength at the nuclear site. The phenomenon is termed shielding. Hence, the more shielded nuclei have lower Larmor frequencies.
It is not convenient to accurately measure the absolute values of transition frequencies, as is done with other spectroscopic procedures. However, it is convenient to accurately measure the difference in frequencies between two resonance signals. The position of a signal in an NMR spectrum is described by its separation from another resonance signal arbitrarily taken as standard. This separation is called chemical shift, which may be expressed in units of magnetic field or in frequency units that are readily interconverted by the equation for the resonance condition, Equation 6. This equation shows that when the separation is expressed in frequency units, it is directly proportional to the field strength. It is more convenient, therefore, to express the chemical shift in terms of the dimensional unit  , which is independent of the field strength, and is defined by
, which is independent of the field strength, and is defined by
where  SS is the test substance resonance frequency, in Hz; RS is the reference resonance frequency in Hz; and 0 is the instrument frequency, in MHz. When 0 is expressed in units of MHz, then Equation 7 is expressed in units as parts per million (ppm). Hence, it is common to use the unit ppm to express the chemical shift difference between the resonance peak of the test substance and that of the reference.
SS is the test substance resonance frequency, in Hz; RS is the reference resonance frequency in Hz; and 0 is the instrument frequency, in MHz. When 0 is expressed in units of MHz, then Equation 7 is expressed in units as parts per million (ppm). Hence, it is common to use the unit ppm to express the chemical shift difference between the resonance peak of the test substance and that of the reference.
By using this equation, one can use (with appropriate caution) the chemical shift of any known species (such as the residual 1H-containing species in a deuterated solvent) as a chemical shift reference. This equation, now in common use, is applicable to nearly all nuclei.
Tetramethylsilane (TMS) is the most widely used chemical shift reference for proton and carbon spectra. It is chemically inert, exhibits only one peak (which is more shielded than most signals), and is volatile, which allows ready specimen recovery. Either 2,2,3,3-d4 sodium 3-(trimethylsilyl)propionate (TMSP) or sodium 2,2-dimethyl-2-silapentane-5-sulfonate (DSS) is used as an NMR reference for aqueous solutions. The resonance frequency of the TMSP or DSS methyl groups closely approximates that of the TMS signal. Where the use of an internal NMR reference material is not desirable, an external reference may be used, such as a reference standard in a separate NMR tube.
Conventional NMR spectra are shown with shielding increasing and chemical shift decreasing from left to right because less shielded nuclei have higher Larmor frequencies than do more shielded nuclei. Resonances on the left are said to be deshielded (i.e., they show lower electron density). Resonance peaks appearing at the right are termed more shielded (i.e., they show greater electron density) than those appearing at the left. Resonances from the more shielded and the less shielded nuclei often are inappropriately called the high-field or upfield peaks and the low-field or downfield peaks, respectively, as a result of the outdated method of acquiring data by sweeping the magnetic field. Today, the overwhelming majority of spectra are acquired on a pulsed Fourier transform (FT) spectrometer, which sweeps neither the magnetic field nor the transmitter frequency. Therefore, it is more appropriate to refer to resonances at the left side of the spectrum as high-frequency or deshielded resonances and those on the right as low-frequency or shielded resonances.
The coupling between two nuclei can be described in terms of the spin-spin coupling constant, J, which is the separation (in Hertz) between the individual peaks of the multiplet. When two nuclei interact and cause reciprocal splitting, the measured coupling constants in the two resulting mutiplets are equal. Furthermore, J is independent of magnetic field strength.
Coupled spin systems are usually referred to as weak or strong. These terms depend on the separation of the Larmor frequencies of the coupled nuclei compared to the coupling constant between them. Both of these values are easily measured from the spectrum. For a weakly coupled system, the separation expressed in Hz (D) is large compared to J, which is always expressed in Hz. Thus, the ratio of the two is dimensionless. For a weakly coupled system, the ratio is large. Typically, spectroscopists consider a ratio above 10 to be weak. Only weakly coupled spin systems produce first-order spectra, which are comparatively easy to analyze. The number of individual peaks that are expected to be present in a multiplet and the relative peak intensities are predictable. The number of peaks is determined by 2 nI + 1, where n is the number of identical nuclei on adjacent groups that are active in splitting and I is the spin of those nuclei causing the splitting. For protons this becomes (n + 1) peaks. In general, the relative intensity of each peak in the multiplet follows the coefficient of the binomial expansion (a + b)n. These coefficients can conveniently be found by use of Pascal’s triangle, which produces the following relative areas for the specified multiplets: doublet, 1:1; triplet, 1:2:1; quartet, 1:3:3:1; quintet, 1:4:6:4:1; sextet, 1:5:10:10:5:1; and septet, 1:6:15:20:15:6:1. Two examples of first-order spectra arising from weak coupling are shown in Figure 1.
+'&img=../../images/v38332/c1761-f1-pf376-new.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 1. Diagrammatic representation of simple spectra resulting from weakly coupled spin systems.
Coupling may occur between 1H and other nuclei, such as 19F, 13C, and 31P. This type of coupling can frequently be observed between nuclei separated by 1–5 bonds.
Magnetically active nuclei with I  1, such as 14N, possess a nuclear quadrupole moment, which produces line broadening of the signals from neighboring nuclei.
1, such as 14N, possess a nuclear quadrupole moment, which produces line broadening of the signals from neighboring nuclei.
Another characteristic of an NMR signal is its relative intensity, which has wide analytical applications. In carefully designed experiments, the area or intensity of a signal is directly proportional to the number of protons that give rise to the signal. As a result, NMR can be used for quantitation (see Quantitative Analysis in this chapter and in Nuclear Magnetic Resonance, Qualitative and Quantitative NMR Analysis  761
761 . NMR spectra may contain spinning side bands, peaks that appear symmetrically located around each signal. These signals are due to the failure to optimize the off-axis (x and y) shims. The homogeneity of modern superconducting magnets, coupled with computer shimming techniques, reduce the need for sample spinning and completely eliminate these sidebands.
. NMR spectra may contain spinning side bands, peaks that appear symmetrically located around each signal. These signals are due to the failure to optimize the off-axis (x and y) shims. The homogeneity of modern superconducting magnets, coupled with computer shimming techniques, reduce the need for sample spinning and completely eliminate these sidebands.
NMR SPECTROMETERS
Introduction
NMR spectrometers have evolved since the first commercial instrument, a Varian HR-30 that operated at 30 MHz, was produced in 1952. Initially NMR spectrometers used a data acquisition technique known as continuous wave (CW), which was based on sweeping the magnetic field. The limitations of a CW spectrometer include low sensitivity and long analysis time.
Today’s spectrometers operate at frequencies up to 1 GHz and apply an rf pulse to the sample to produce a time-domain signal known as a free induction decay (FID), which is then Fourier transformed into a frequency-domain signal. This technique is known as FT NMR spectroscopy. Current NMR spectrometers are composed of several key components: the magnet, the probe, the console, and the computer.
The instruments are described by the approximate resonance frequency of the 1H resonance, e.g., 600 MHz, or by their field strength, e.g., 14.1 T.
The Magnet
Until the early 1970s, NMR magnets were either ferromagnetic-core electromagnets or permanent magnets that operated at field strengths of 1.41–2.35 T, corresponding to 1H resonance frequencies of 60, 80, 90, and 100 MHz. The first NMR magnets based on superconducting magnets were introduced in the 1960s and allowed access to much higher field strengths that currently are as high as 23.5 T (1 GHz).
The superconducting magnet is the most expensive component of an NMR spectrometer and can cost up to several million dollars. These magnets consist of miles of Nb3Sn or NbTi wire. When these materials are wound into a solenoid that is immersed in liquid helium at 4.2 K, they become superconducting. That is, an electrical current can be induced in them by an energizing power supply, and that current will persist for many years even after the power supply is removed. This essentially constant electrical current is used to generate high static magnetic fields that can be several times higher than those obtained with ferromagnetic-core magnets. Ensuring that the superconducting coil is immersed in liquid helium at all times is the only maintenance needed to sustain these high fields.
Figure 2 contains a diagram of a typical superconducting magnet. The superconducting solenoid is immersed in a liquid-helium Dewar at 4.2 K. This unit is itself contained in a liquid-nitrogen Dewar at 77.4 K. Each Dewar is surrounded by a vacuumed space and reflective film coating to prevent outside heat of the laboratory from entering the helium Dewar. The central core or room-temperature bore provides room for the stack that contains the shim coils for room-temperature shimming. Finally the probe is inserted into the stack. Samples are injected or ejected from the probe by means of a jet of filtered and dried air or nitrogen.
+'&img=../../images/v38332/c1761-f2-pf376-new.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 2. Schematic representation of a superconducting magnet.
An important recent advance in magnet technology has been the construction of shielded magnets with stray fields that can extend only one or two meters from the center of the magnet in three dimensions, thereby making magnet siting a far easier task than it was with unshielded magnets.
In addition to the main solenoid, the helium Dewar also contains several other superconducting coils that are used to shim the main magnetic field as a first step in attaining very high field homogeneity. Further shimming is accomplished by 20–30 shim coils in the room-temperature shim stack that is inserted in the bore of the magnet. These coils operate at ambient temperature and are used to generate small magnetic fields that oppose and cancel inhomogeneities caused by the surroundings, the probe, or the sample itself. Computer software has taken over a large amount of the tedious job of shimming the magnet homogeneity, a critical task for obtaining good NMR data. Using the lock signal from the sample, spectroscopists can generate a field map for each of the shim coils. Using this map, the computer then calculates the amount of current that should be applied to each of the shim coils to maximize the magnet homogeneity. Typically this operation takes less than a minute for shimming the on-axis (z1 to z6) shim coils. The off-axis (x/y) inhomogeneities can be compensated for in a similar manner but usually in a longer period of time (15–20 minutes) because of the larger number of off-axis coils.
The Probe
The NMR probe may be the most important part of the instrument. The probe consists of one or two rf coils. Each coil, which is inductive (L), is in a circuit that contains several tunable capacitive (C) elements. These elements are adjusted to enable the probe to transmit and receive at the Larmor frequency, 0. For a given nucleus, the probe tuning is determined by [ = 1/(2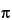 (LC)1/2)]. A pulse of rf at the Larmor frequency results in an applied magnetic field (B1). To induce a transition, B1 must be applied perpendicular to the static field generated by the magnet superconducting coil. The rf coil not only transmits the excitation pulse but is also electronically switched to receive the rf signal from the sample.
(LC)1/2)]. A pulse of rf at the Larmor frequency results in an applied magnetic field (B1). To induce a transition, B1 must be applied perpendicular to the static field generated by the magnet superconducting coil. The rf coil not only transmits the excitation pulse but is also electronically switched to receive the rf signal from the sample.
The most common NMR sample tube is the 5-mm (od) NMR tube. However, probes have been designed in many forms. Some probes can accommodate 10- or 20-mm tubes for samples that are not in limited supply, such as petroleum and polymers. For limited amounts of precious samples, probes have been made to accept tubes as small as 1 mm, which can accommodate solutions as small as 5 µL. Also, flow probes are available to obtain data directly from a liquid chromatography effluent.
Probes are available in a large number of coil configurations. The most common probe usually contains a broadband observe coil that is tunable over a wide range of frequencies (31P to 109Ag), a decoupler (1H) coil, and a coil tuned to deuterium (2H) for field-frequency lock. Usually, the decoupler coil is double-tuned for both 1H and 2H. Probe configurations can include as many as four channels for multidimensional work with biological macromolecules.
The probe can come in a format where the X-nucleus observe-coil (e.g., 13C) is wound closest to the sample for highest sensitivity, and the decoupler coil (1H) is farther away. The inverse configuration is also available to provide the maximum signal-to-noise (S/N) ratio for 1H, the detected nucleus in two-dimensional (2-D) indirect heteronuclear experiments, such as heteronuclear single-quantum coherence spectroscopy (HSQC) and heteronuclear multiple-bond correlation (HMBC) where the X-nucleus is indirectly detected via the 1H frequency. Recent advances in probe technology have resulted in a single probe combining the sensitivity of the direct and inverse detection probes described above. Another advancement in probe design is the cryogenically cooled probe wherein the rf coils and their preamplifiers are held at close to liquid helium temperatures (20 K). Because rf electronics generate lower noise levels at colder temperatures, S/N ratios can be increased by at least a factor of four in these probes. Because the S/N ratio of a spectrum is given by n1/2, where n is the number of acquisitions, an enhancement by a factor of four to the initial S/N ratio translates to a savings of 16 in time or to a four-fold reduction in sample size.
Probes can also be equipped with gradient coils that can apply a magnetic field gradient in the z only or x, y, and z directions. These gradients are used to study diffusion or, more commonly, to be integral parts of pulse sequences because they provide an efficient means for selecting specific coherences in 2-D experiments.
In addition to the electronic coils, probes usually come equipped with a heater coil that enables variable temperature work from 100 to +150. Probes also have gas outlets to allow sample insertion and ejection from the probe as well as sample spinning.
to +150. Probes also have gas outlets to allow sample insertion and ejection from the probe as well as sample spinning.
For solid-state samples the probes come with cylindrical rotors that are filled with the sample and capped. The entire rotor can then be oriented at an angle of 54.74 (known as the magic angle) relative to the magnetic field direction and spun at rates up to 70 kHz. Rapid rotation at this angle helps remove the chemical shift anisotropy and dipolar coupling so that the very broad resonances observed in the solid state can be reduced considerably.
The Console and Computer
The NMR console has the primary function of generating the various Larmor frequencies required for a given experiment, amplifying and transmitting these frequencies to the probe, and detecting the resulting signals that are transmitted from the probe so that they can be used to create an NMR spectrum. In addition to these primary functions, the console performs many more operations, all of which are computer-controlled. Figure 3 contains a schematic diagram illustrating the various components of the current majority of spectrometer consoles.
+'&img=../../images/v38332/c1761-f3.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 3. Schematic diagram of an NMR spectrometer.
The computer continually controls the signal to the lock transmitter (A) so the resonance of the lock material can be detected in the lock receiver to maintain field/frequency control. The computer also controls the frequency synthesizer (B) that generates the various frequencies that are close to the Larmor frequencies of the observed or perturbed nuclei. For a specific experiment, the computer triggers the pulse programmer (C), which, in turn, sends timing signals (pulse width, phase, and shape) to the observer transmitter (D), the decoupler (E), and the gradient amplifier (F), depending on which of these units are needed for that experiment. Once the observed nucleus in the probe (G) is excited by a pulse from the observed transmitter, the resultant rf signal is amplified in the preamplifier (H). Then the signal is mixed (J) with a local oscillator (I) to generate a lower rf frequency, called the intermediate frequency (IF) that is further amplified (K). A second mixing stage, this time with a different local oscillator (L), results in an audio signal that is then detected in quadrature (M) before being amplified (N), converted (O) from an analog to a digital signal, and stored in the computer. After the signal is processed to create a spectrum, the digital data can be converted back to an analog signal in a digital-to-analog converter (P) and printed.
The output from the phase-sensitive detector (M) is the free induction decay (FID), which is a time-domain signal, f(t).
When two detectors are used (M) with their reference frequencies shifted from each other by 90, frequencies that are positive with respect to the reference can be distinguished from those that are negative. This system is referred to as quadrature phase-sensitive detection (QPD). Each detector produces an FID, but they will always be 90 out of phase with respect to each other. One FID is called the real FID, and the other is called the imaginary FID. The Fourier transform that is performed in this case is called a complex Fourier transform. The combination of the real and imaginary FIDs yields the frequency spectrum, F().
+'&img=../../images/v38332/c1761-eq8-pf376-new.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
In addition to the Fourier transform process, the computer is also used for post-acquisition processing of the data. The frequency-domain spectrum that results from the complex Fourier transform can then be phased, baseline corrected to remove distortions, integrated to obtain peak areas, and peak picked to provide chemical shift information. The computer is also capable of providing spectra calculated from chemical shift and coupling values, curve-fitting resonances, and deconvoluting complex overlapped peaks. Finally, the digital data can be converted to their analog form by a DAC (O) and printed.
RELAXATION
NMR includes two types of relaxation: Spin-Spin Relaxation, sometimes referred to as Transverse Relaxation, or T2 Relaxation, and Spin-Lattice Relaxation, sometimes referred to as Longitudinal Relaxation, or T1 Relaxation. At least two mechanisms contribute to Transverse Relaxation: loss of signal due to B0 inhomogeneity and the natural relaxation that would take place even in a perfectly homogeneous field. The combined effects of these two mechanisms produce a new time constant for the relaxation, which is referred to as T2*.
Spin-Spin Relaxation (Transverse Relaxation)
After an rf pulse, the component of M0 in the (x,y) plane, Mxy, will gradually decay toward zero. The process is first order, and as it is in other types of first-order processes, the instantaneous rate of decay of Mxy is directly proportional to its displacement from equilibrium. The further Mxy is displaced from zero, the faster it decays, and as it approaches zero it decays more and more slowly. Hence, Equation 9 applies.
dMxy/dt 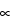 (–Mxy) [9]
(–Mxy) [9]
The process is analogous to the decay of a radioactive element. However, spectroscopists do not speak of the rate of decay of an FID in terms of its half-life. Instead, they speak of its 1/e-life, that is, how long it takes for the FID to decay to a value that equals 1/e of its original value at time zero. Standard mathematical manipulations of Equation 9 yield
Mxy = M0exp(–t/T2*) [10]
where M0 is the equilibrium distribution given by Equation 9, and T2* is the rate constant for the decay. T2* is a measure of how fast the signal decays, not how long it takes to decay. The rate of decay is fastest immediately after the pulse because then it is furthest from its equilibrium position, zero. If the rate remained constant at this initial rate, then the signal would be completely decayed after one T2*.
Table 3. Percent of Mxy Remaining as a Function of Time, in Units of T2*
| Time/T2* | 0.0 | 0.5 | 1.0 | 1.5 | 2.0 | 3.0 | 4.0 | 5.0 |
| % Remaining | 100.0 | 60.7 | 36.8 | 22.3 | 13.5 | 5.0 | 1.8 | 0.7 |
Mxy asymptotically approaches zero, and it would take infinitely long for complete decay, but the decay is normally considered to be complete when the time has reached 3 to 5 times T2*. Hence, these times are commonly used as acquisition times. If acquisition times that are shorter than 3 times T2* are used, the FID is truncated and subsequent Fourier transform leads to visible baseline artifacts.
The decay of the FID produces the peak width in the final spectrum. The faster the decay, the broader is the line. The mathematical relationship is:
D1/2 = 1/(T2*) [11]
where D1/2 is the width of the peak at its half-height.
Spin-Lattice Relaxation (Longitudinal Relaxation)
After an rf pulse, nuclei are excited from the low-energy state into the high-energy state. The nuclei will eventually relax back to establish the Boltzmann distribution (see Equation 5), and this process is called spin-lattice relaxation. The recovery process is first-order, and as it is in other types of first-order processes, the instantaneous rate of growth of Mz is directly proportional to its displacement from equilibrium. The farther Mz is displaced from M0, the faster it grows back, and as it approaches M0 it grows back more and more slowly. Hence, Equation 12 applies.
dMz/dt (M0 Mz) [12]
Standard mathematical manipulations yield
Mz = M0(1 – exp(– t/T1) [13]
Note that T1 is a measure of how fast Mz grows back to M0—it is not how long it takes to grow back. Table 4 was prepared from Equation 13 and shows the percentage of recovery of Mz as a function of time after a 90 pulse. In this table, time is given as the number of T1s.
Table 4. Percent Recovery of Mz as a Function of Time, in Units of T1
| Time/T1 | 0.0 | 0.5 | 1.0 | 1.5 | 2.0 | 3.0 | 4.0 | 5.0 |
| % Recovery | 0.0 | 39.3 | 63.2 | 77.7 | 86.5 | 95 | 98.2 | 99.3 |
As Mz asymptotically approaches M0, it takes infinitely long for 100% recovery. However, recovery is normally considered to be complete when it has reached 99%. Hence, relaxation delays of 5T1 are commonly used in pulse sequences. The rate of return to M0 is fastest immediately after the pulse because that is when M0 is farthest from its equilibrium position. If the rate remained constant at this initial rate, then full recovery would be achieved after one T1.
TIP ANGLE
During an rf pulse, a magnetic field (B1) is applied to the sample. The magnetization vector M precesses about B1 according to
where 1 is the precessional frequency and B1 is the strength of the magnetic field applied to the sample. During the time that the pulse is applied, M precesses to an angle ( ) given by the precessional rate multiplied by the width of the pulse, in time, (PW). Then,
) given by the precessional rate multiplied by the width of the pulse, in time, (PW). Then,
PW × 1 = = PW × B1 [15]
Tip angles typically are expressed in units of degrees or in radians. For example, a 90 pulse is sometimes referred to as a /2 pulse.
Optimum Tip Angle or Ernst Angle
Time averaging to improve the S/N ratio is accomplished by a delay-pulse-acquire sequence that is repeated as necessary. Suppose that during the first pulse, M0 precesses 30 about B1 before the pulse is turned off. At this time the magnitude of Mz is M0cos 30. At the end of the pulse, Mz will begin to grow back toward its equilibrium value, M0. Typically, the second pulse is applied before Mz reaches M0, driving Mz down even farther away from M0. Consequently, Mz begins to grow back faster because it is even farther away from its equilibrium value. After 6–10 pulses, Mz will grow back an amount that is equal to the incremental displacement caused by each succeeding pulse and will reach a new equilibrium or steady state. Each succeeding pulse continues to tip this new steady-state value of Mz by 30, resulting in signal intensity for each pulse.
The position of the steady-state equilibrium is determined by three factors: the T1, the tip angle, and the time between pulses. On the one hand, for a given T1 and time between pulses, if the tip angle is too large then the steady-state value of Mz will be close to the origin, affording only a small signal. On the other hand, if the tip angle is too small, e.g., 5, then the steady-state value will be large, but with a small angle the value of Mzsin 5 will also be small, again affording a small signal. The optimum angle is frequently called the Ernst angle, which is given by:
cos opt = exp(PR/T1)[16]
where PR is the time between pulses, or pulse-repetition time. This time is the sum of the acquisition time used to collect the FID plus any relaxation delay used. This angle provides a reasonably large steady-state value combined with a reasonably large angle and will produce the best S/N ratio per unit time. The value of T1 to be used in Equation 17 should be for the longest relaxing nucleus in the molecule.
RELAXATION DELAY
Surprisingly, the optimum S/N ratio per unit time is obtained when no relaxation delay is used for a given T1. That is when PR equals to AT, the acquisition time. However, because the nuclei in most molecules do not have the same T1 values, the relative intensity relationship will be lost.
For typical quantitation experiments, a relaxation delay is used and should be at least five times the longest T1 expected for any of the nuclei in the molecule; and the pulse width should be set to 90. Further details are provided in the section Quantitative Applications.
RESOLUTION
In NMR spectroscopy the typical definition of resolution is the ability to distinguish between two closely spaced resonance peaks in a spectrum. The industry standard for measuring resolution is to measure the width of a single peak, in units of Hz, at the half-height of the peak.
The uncertainty principle determines the best resolution that can be achieved in an NMR spectrum. The maximum resolution possible, or the minimum separation that can be observed between two frequencies, D, in a spectrum is equal to the reciprocal of the acquisition time, AT, of the FID.
D = 1/Dt = 1/AT [18]
The time set by the spectrometer operator should not exceed the required AT, because this would result in the collection of only noise after the signal has decayed to near zero. Collecting this noise does not improve the resolution.
POSTACQUISITION DATA PROCESSING
The final appearance of the spectrum can usually be improved by applying a variety of mathematical procedures to the FID before the Fourier transform is performed. The two most common procedures are multiplying the FID by a mathematical function, generally known as a window function; or appending zeros to the end of the FID, generally known as zero filling.
Window Functions
Two types are generally used: one for increasing the resolution and another for increasing the S/N ratio.
increasing the resolution
The decay of the signal produces a peak width in the spectrum, and if this decay can be removed, then the resonance peak would consist of a single point, i.e., an infinitely narrow peak. The decay of the signal can be represented by exp( t/T2*). Hence the full equation representing the decaying signal is
A(t) = A0exp( t/T2*)cos(t + 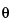 ) [19]
) [19]
If the FID is multiplied by an increasing function that exactly cancels the decay, then the peak width will be removed. This can be achieved by multiplying the FID by exp(t/T2*). Then Equation 19 becomes
A(t) = A0exp(0)cos(t + ) = A0cos(t + ) [20]
Unfortunately, the application of this function as described above will also disproportionately increase the noise power at the tail of the FID. The S/N ratio in the final spectrum is so poor that this function is not used without modifications. Typically, the beginning of the FID, where the S/N ratio is better, is multiplied up but then the tail of the FID, where S/N ratio is poorer, is multiplied down. Two commonly used functions that accomplish this are the Gaussian function and the transform of reversed-added FIDs (TRAF) function for resolution. The latter is sometimes given the name TRAFR by instrument manufacturers. It affords the same resolution enhancement as the Gaussian function but with much less degradation in overall S/N ratio. Numerous other window functions have been proposed though not always widely used. It should also be noted that in the quantitative experiments increasing the resolution should be used with caution because it may change the accuracy of signal integration in the spectrum.
increasing the s/n ratio
The overall S/N ratio in the spectrum can be increased by weighting the points at the beginning of the FID more highly than at the tail. This is because the S/N ratio is the highest in the beginning and the lowest at the tail. The weighting is often accomplished by multiplying the raw FID by a function that decreases with time. A popular function that gives the greatest increase in the S/N ratio is called the matched filter. It weights each point in the FID by an amount proportional to the S/N ratio at that point. A function that accomplishes this must match the decay. Hence, the FID is multiplied by exp( t/T2*).
The penalty for the use of the matched filter is a loss of resolution that equals a doubling of the peak width. When the original decay is multiplied by the matched filter, then the new decay is given by the following:
exp( t/T2*) × exp( t/T2*) = exp( 2t/T2*) = exp( t/0.5T2*)[21]
The FID then appears to have decayed with a T2* equal to one-half of the original, and according to Equation 11 and 18 the peak width will double. Multiplying the FID by a steeper decay in an attempt to weight the beginning points eore results only in less of an increase in S/N ratio and a greater increase in peak width.
Another function that accomplishes the same increase in S/N ratio, but without any change in resolution, is the TRAF function for sensitivity, which is sometimes given the name TRAFS by instrument manufacturers.
Zero Filling
Spectroscopists can improve the overall appearance of the spectrum considerably by appending zeros to the end of the FID before the Fourier transform is performed. This process results in placing more points on every resonance peak in the spectrum. The most common procedure is to append a number of zeros equal to the number used to collect the FID. Adding more zeros will result in only a very slight further improvement.
Although all of the peaks are better defined with zero filling, the resolution is not increased. For example, in a case where the separation between two lines is closer than the peak widths, only a single broadened line will result. Zero filling will not resolve these peaks—it will only place more points on an already broadened line. Only an increase in the acquisition time of the signal or the use of a window function could resolve the peaks.
Zero filling has a beneficial effect on quantitation. Integration of a digital spectrum is accomplished by taking the intensity at a given point and adding to it the intensity at successive points. If the number of data points is insufficient to depict the actual peak shape, the resultant integral of that peak will not be accurately determined. Therefore, zero filling until each peak is represented by at least 7–10 data points results in a more accurate integration. To obtain reliable peak representation and quantitative peak integration there should be at least 4–5 data points above the full width at half height of a peak.
GENERAL PROCEDURE FOR STRUCTURE IDENTIFICATION
NMR spectroscopy is a powerful technique for structure identification because of its specificity of detecting certain nuclei such as 1H, 13C, 31P, and 19F. Typically, a routine identification test can be performed by 1H NMR spectroscopy in a short period of time for simple molecules. The basis for identification is provided by a comparison of the signals from the test sample with the expected signals from a qualified reference standard. A positive identification can be concluded when the chemical shifts, multiplicities, and coupling constants of the spectrum of the test sample match those of the reference standard or, in the case of a USP monograph, the values listed in the monograph.
Data may be made unacceptable for analysis if incorrect sample preparation or poor adjustment of spectrometer parameters leads to poor resolution, decreased sensitivity, and spectral artifacts. It is preferable that the operator be familiar with the basic theory of NMR and operation of the spectrometer. Frequent checks of instrument performance are essential.
The procedures discussed here refer specifically to 1H and 13C NMR, but they are applicable, with modification, to other nuclei. The discussion assumes that the NMR spectra are obtained from solutions in suitable solvents.
Selection of Solvent
Deuterated solvents are usually used to prepare solutions for NMR analysis because they are readily available, have greatly reduced 1H signals from solvents in 1H spectra, and have the added advantage of providing a lock signal. Select a solvent whose residual 1H signals will not interfere with signals of the analyte. If a residual 1H solvent peak might interfere with any signals from the sample solution and another solvent is not possible, then the 2H isotopic purity of the solvent should be as high as possible. Some solvents (e.g., D2O or CD3OD) have labile protons that can enter into fast exchange reactions with the labile protons in the analyte. This may eliminate resonance signals from –COOH, –OH, and –NH2 structural groups. The most commonly used solvents for proton and carbon NMR are listed in Table 5 along with their residual 1H and 13C chemical shifts as well as the multiplicities of these resonances caused by coupling to deuterium.
Table 5. Solvents Commonly Used for 1H/13C NMR Chemical Shifts
| Solvent | Residual 1H/13C Signal ( and Multiplicity |
|
|---|---|---|
| 1H | 13C | |
| CDCl3 | 7.27 | 77.23 (3) |
| CD3OD | 3.35, 4.78 | 49.15 (7) |
| (CD3)2CO | 2.05 | 206.68 (1) 29.92 (7) |
| D2O | 4.7c | — |
| (CD3)2SO | 2.50 | 39.51 (7) |
| C6D6 | 7.20 | 128.39 (3) |
| Dioxane-d8 | 3.55 | 66.66 (5) |
| CD3CO2D | 2.05, 11.65c | 178.99 (1) 20.0 (7) |
| (CD3)2NCDO | 2.77, 2.93, 8.05 | 163.15 (3) 34.89 (7) 29.76 (7) |
|
a
Chemical shifts were measured at 295 K>.
b
c
Labile hydrogen.
|
||
Sample Preparation
For USP procedures, directions are usually given in individual monographs. The solute concentration depends on the objective of the experiment. Typically, NMR sample solutions are prepared so that they contain from a few to 50 mg/mL. Detection of minor contaminants may require higher concentrations. In some cases such as polymers, even higher concentrations can be used. The solutions are prepared in separate vials and are transferred to the NMR tube. The volume required depends on the size of the NMR tube and on the geometry of the probe. The level of the solution in the tube must be high enough to extend beyond the coils when the tube is inserted in the instrument probe.
The NMR tubes must meet narrow tolerance specifications in diameter, wall thickness, concentricity, and camber. The most widely used tubes have a 5- or 10-mm outside diameter (OD) and a length between 15 and 20 cm, but 1- and 3-mm (OD) NMR tubes are becoming more common, and tubes as large as 20 mm (OD) have been used.
Procedure
The NMR tube is placed in a probe located in the magnetic field. Although samples traditionally have been spun to average the nonradial field gradients, the quality of the shim coils no longer makes spinning a requirement, and, in the case of many 2-D experiments, the sample should not be spun. The magnetic field’s homogeneity is optimized by shimming, a function that is largely being taken over by the computer in most modern spectrometers. Probe tuning is optimized for the frequency being observed and is matched to the impedance of the spectrometer.
The computer serves to control all operations of the spectrometer from running the pulse program to storing and processing the data. The experimental setup involves selecting values for a large number of variables, including the spectral width to be examined, the duration of the excitation pulse (PW), the time interval over which data will be acquired (AT), the number of transients to be accumulated, and the delay between one acquisition and the next (relaxation delay). The acquisition time for one transient is on the order of seconds. The number of transients is a function of the specimen concentration, the type of nucleus, and the objective of the experiment and can vary from a few for most 1H experiments to several thousand for 13C spectra. At the end of the experiment, the signal (FID) is stored in digitized form in the computer memory and may be displayed on the monitor. The signal can be processed mathematically to enhance either the resolution or the sensitivity, and it can be Fourier-transformed into a frequency-domain spectrum, which can be further analyzed to obtain peak positions (chemical shifts) and intensities.
Structure Elucidation by NMR
The simplest case of using NMR spectroscopy to elucidate an unknown structure is to obtain a match with a spectrum from a reference standard or from a database. The informational content of an NMR spectrum is sufficient for deducing structures of organic molecules even when qualified reference standards or spectra are not available. Relatively simple structures can be identified using chemical shifts, coupling patterns, and intensities obtained from one-dimensional (1-D) 1H and 13C spectra. For more complex structures, spectroscopists may have to obtain two-dimensional (2-D) spectra from experiments that have been developed to determine homo- or heteronuclear connectivities.
Two-dimensional spectra are characterized by two frequency axes. The intensity, which is mathematically another dimension, is not considered to be a dimension in 2-D NMR because it is not an axis that spreads out the chemical shift. All modern 2-D experiments consist of at least two pulses separated by a time period, labeled t1, the evolution period, and a period of time used for collecting the signals, labeled t2, the detection period. A COrrelation SpectroscopY (COSY) sequence is the simplest of all and is shown in Figure 4.
+'&img=../../images/v38332/c1761_fig4_usp36.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 4. Pulse sequence for a COSY experiment.
A series of such sequences is performed using an incremental increase in the evolution period. Fourier transforms of the FIDs produced during each of the t2 detection periods are stacked, a second Fourier transform is performed along this t1 new axis, resulting in a plot of amplitude along two frequency axes, F2 and F1.
The next simplest addition to the COSY sequence is another pulse. The Nuclear Overhauser Effect SpectroscopY (NOESY) sequence is an example and is shown in Figure 5.
+'&img=../../images/v38332/c1761_fig5_usp36.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 5. Pulse sequence for a NOESY experiment.
The evolution period is again the time between the first two pulses and is incremented as in the COSY experiment. The time between the second and third pulses is fixed and is not incremented. This period of time is labeled 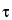 m and is called the mixing time. Cross-polarization occurs during this period.
m and is called the mixing time. Cross-polarization occurs during this period.
The COSY and NOESY sequences described above are the two simplest 2-D experiments to perform. Many other experiments have been developed and contain a very complicated series of pulses with different pulse widths, time delays, gated decoupling, and pulsed field gradients. Some of these experiments are described below.
Strategies for Establishment of Homonuclear Connectivities
Assigning signals based on chemical shifts, spin multiplicity, and coupling constants serves as a starting point for structural elucidation. Structure elucidation is simplified if one can establish molecular connectivity between homonuclear spins. This can be done using correlations via bonds (scalar coupling, sometimes referred to as J coupling) or via spatial (dipolar coupling) interactions. This section describes NMR techniques that can be used for the study of homonuclear connectivity. Popular experiments in this category are COSY, total correlated spectroscopy (TOCSY), NOESY, and rotating frame Overhauser effect spectroscopy (ROESY).
cosy
COSY has become a routine 2-D 1H NMR experiment that can quickly provide the proton–proton connectivity for spin systems connected by two or three chemical bonds. In a COSY spectrum, the contour plot typically shows diagonal and off-diagonal cross-peaks. The diagonal peaks correspond to the places in 2-D space where chemical shifts of the same nucleus in the 1-D 1H NMR spectrum intersect. The off-diagonal cross-peaks occur in 2-D space where the chemical shift of one nucleus intersects the chemical shift of a different nucleus to which it is coupled. The COSY spectrum enables the identification of scalar-coupled spins that are in geminal or vicinal positions. Generally, one starts the assignment process by selecting a resonance in the COSY spectrum that has already been identified in the 1-D 1H NMR spectrum. Then, the off-diagonal peaks between this resonance and any others determine the resonances of the neighboring protons to which it is coupled. The neighboring protons identified in this way then serve as the next points to examine for cross-peaks to other protons, and so forth. The process continues until all coupled spin systems are identified.
Thus, a COSY experiment can provide useful information about proton–proton connectivity for various fragments of the molecules under examination. The relationship between these fragments may be difficult to establish because of an interruption of the coupling between the two fragments. For instance, if two segments are connected through quaternary carbons or heteronuclei, the very small four-bond coupling between two protons on the two separate fragments usually cannot be detected by COSY. Basic COSY experiments are usually processed using magnitude mode, which results in broad bases to the peaks. The diagonal peaks may be so broad that spin systems with close chemical shifts may not be observable because the broadness of the diagonal peaks will cause them to overlap the off-diagonal peaks. An example of a COSY experiment, specifically a gradient, or gCOSY, is shown in Figure 6.
+'&img=../../images/v38332/c1761-f6-pf376-new.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 6. Partial COSY spectrum of sucrose. Dotted lines show how two off-diagonal (f3, f4 and g2, g3) contours are used to determine which nuclei are coupled to each other.
A variety of COSY experiments have been devised to improve upon the original experiment. Perhaps the most important change has been to use gradients to acquire a gCOSY spectrum. The classic COSY experiment uses a 90 pulse to generate transverse magnetization and relies on elaborate phase cycling to cancel unwanted signals over many scans, resulting in long experiment times. In a gCOSY experiment, which requires a probe capable of a pulsed field gradient (PFG), a magnetic field gradient pulse dephases any coherent magnetization in the xy plane. If a second gradient pulse of the proper strength is applied in the opposite direction, it will cause any dephased double-quantum magnetization to refocus. Hence, only those signals will be received. The strengths of the gradients can be used to select single-, double- or triple-quantum coherences.
The gradient pulses in a gCOSY experiment can be used to prevent the refocusing of magnetizations that cause the artifacts in the classic COSY experiment. Hence, instead of requiring a minimum of eight phase-cycled acquisitions for each data increment in the second dimension, a gCOSY spectrum requires only a single acquisition per increment. For samples that are sufficiently concentrated to produce an acceptable S/N in only one acquisition, this greatly shortens the experimental time.
Phase-sensitive COSY experiments have been developed to overcome the problem of overlap with closely spaced chemical shifts. A phase-sensitive COSY results in pure absorptive peaks with narrower peaks than are generated by the classic-magnitude COSY. These narrower peaks allow better resolution of resonances that are close to diagonal peaks.
Double-quantum filtered COSY (DQF-COSY) was developed to overcome the problem caused by intense signals from functional groups such as methyls. The singlets from these groups do not provide useful connectivity information, but their intensity often limits the dynamic range of the experiment, making it difficult to observe other weaker signals. The pulse sequences of DQF-COSY detect the spin systems that have only double-quantum transitions. Isolated singlets are not selected and thus are filtered out of the final 2-D spectrum. In addition, a reduction in the overall intensity of the diagonal signals is achieved with an increase in the intensities of off-diagonal signals.
tocsy (or hohaha—homonuclear hartmann hahn)
The 1H–1H TOCSY experiment is closely related to COSY but differs because it yields correlations for every spin in a coupled network. This is especially useful when multiplets overlap or there is extensive strong coupling. For example, consider the network –CHa–CHb–CHc–CHd–CHe–, where each CHn stands for a spin that is coupled through three bonds to the adjacent spin. A COSY spectrum would show correlations for each adjacent pair of hydrogens. On the one hand, the Hb resonance would show connectivities to Ha and Hc but not to Hd. A partial correlation is revealed for each CHn. On the other hand, a TOCSY spectrum would show all off-diagonal contours for every spin in this network. That is, for every peak in this coupling network there would be off-diagonal contours corresponding to CHa, CHb, CHc, CHd, and CHe. Thus, a TOSCY spectrum, such as the one shown in Figure 7, affirmatively identifies all of the spins within the same coupling network. This pattern is easily recognized, especially when there is extensive overlap with other coupled networks. However, a TOSCY experiment cannot establish connectivity between separate networks that are interrupted by heteronuclear atoms, quaternary carbons, or a carbon bearing only exchangeable protons. A TOCSY experiment is useful for the study of large molecules with many separated coupling networks such as peptides, proteins, oligosaccharides, and polysaccharides.
+'&img=../../images/v38332/c1761-f7-pf376-new.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 7. Partial TOCSY spectrum of sucrose showing two sets of contours corresponding to all the coupled nuclei in the glucose and fructose rings in sucrose. The contour for the f1 resonance falls within the glucose spin system and has no coupling.
noesy
The NOESY experiment gives correlations between protons that are close to each other in space even though they may not be connected by bonds. These through-space correlations are made via spin-lattice relaxation. Dipole interactions between protons close in space generate NOE transfers, and the magnetization is aligned along the z axis (B0), producing positive or negative intensity changes that yield cross-peaks that are not normally observable in a COSY spectrum. The sign of the NOESY peaks depends on the size and mobility of the molecule under study. Used in combination with other techniques, a NOESY experiment can establish spatial relations for particular spins and can provide critical information about ring structures and conformations.
1h–1h roesy
The ROESY experiment is similar to a NOESY experiment insofar as it also provides correlations between protons that are close to each other in space, whether or not they are connected via bonds. A ROESY spectrum yields through-space correlations via spin-spin relaxation in the rotating frame. The ROESY experiment utilizes a spin–lock sequence as a mixing time during which NOE transfer occurs among all components of the spins locked in the xy plane. In contrast to a NOESY experiment, in which NOE transfer occurs while magnetizations are aligned along the z axis (B0) producing positive or negative intensity changes, the ROESY experiment depends on NOE transfers occurring in the rotating frame under the influence of a B1 magnetic field. This always results in positive signals no matter how large the molecule or whether its motion is fast or slow. Therefore, a ROESY experiment frequently will provide through-space correlations when the same correlations in a NOESY experiment cannot be detected because of molecule mobility.
incredible natural abundance double quantum transfer experiment
The Incredible Natural Abundance Double Quantum Transfer Experiment (INADEQUATE) uses a double quantum coherence to provide information about 13C nuclei directly coupled to other 13C nuclei. Thus it provides the same sort of information that is available using a COSY experiment for proton couplings. Because of the low probability of two 13C nuclei being attached to each other (only 1 in 10,000 molecules), this technique is usually one of last resort. The ever-increasing sensitivity of cryogenically cooled probes used in modern instrumentation makes this experiment practical in some cases.
Strategies for Establishment of Heteronuclear Connectivities
Although homonuclear 1H–1H connectivity is one important aspect of structure elucidation of organic molecules, the establishment of heteronuclear connectivities is equally important, although somewhat more difficult to obtain given the lower abundance of most heteroatoms. If one has partial assignments in either the 1H or 13C spectrum, the knowledge of this connectivity leads to a much fuller assignment of both spectra. Heteronuclear 2-D spectra do not exhibit a diagonal as is seen in homonuclear correlations. Rather, cross-peaks occur at the point of intersection of the 1H and 13C chemical shifts in the 2-D space as shown in Figure 8.
+'&img=../../images/v38332/c1761-f8-pf376-new.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 8. Partial 1H/13C HSQC spectrum of sucrose showing contours that indicate the one-bond correlation between a given hydrogen and the carbon to which it is attached.
Heteronuclear 2-D spectra are designed so that 1H is the detected nucleus and are usually acquired using inverse-detection probes because the 1H coil is wound closer to the sample than the broadband coil. This results in a better filling factor and a greater sensitivity for the 1H coil. It should be noted that a newer configuration of coils has been developed that provides the same sensitivity but does not utilize the inverse-detection coil arrangement. Typical heteronuclear 2-D experiments include Heteronuclear Single Quantum Coherence (HSQC), Heteronuclear Multiple Quantum Coherence (HMQC), and Heteronuclear Multiple Bond Correlation (HMBC) experiments.
hmqc
The 2-D HMQC experiment provides information about correlation between protons and their attached heteronuclei via the heteronuclear scalar coupling. The sequence selects double quantum coherence transfer between scalar-coupled spins (13C–1H).
hsqc
HSQC spectroscopy is also an inverse chemical shift correlation experiment that yields the same information as HMQC, i.e., the identification of directly bonded hydrogen–carbon interactions. The correlation between heteronuclei is detected via the selection of single quantum coherence transfer using the insensitive nuclear enhancement by polarization transfer (INEPT) sequences. The main advantage of using this sequence instead of the HMQC sequence is that the F1 domain does not contain any proton–proton couplings. Hence, the resolution is improved.
An interesting modification of this sequence is an edited HSQC experiment. This is a phase-sensitive experiment that not only gives one-bond correlations between hydrogen and carbon but also gives methyl and methine correlation peaks that are 180 out of phase with methylene resonances.
hmbc
HMBC spectroscopy is a modified version of HSQC and is suitable for determining long-range (> 1-bond) 1H–13C connectivity. Long-range heteronuclear correlation spectroscopy can yield signals for those nuclei that are separated by 2–4 bonds. This experiment, in conjunction with the other 2-D experiments discussed above, allows one to define the structure of a molecule in great detail.
QUANTITATIVE APPLICATIONS
NMR is one of the most useful techniques for quantitative analysis in chemistry. If appropriate experimental conditions are chosen, the relative intensities of resonances are proportional to the population of the nuclei causing those resonances. NMR experiments can be designed for relative or absolute quantitation, either with an internal standard or without one.
Experimental Design for Quantitative NMR
Design for quantitation involves the elimination or precise measurement of differences in intensities due to spin-lattice relaxation and NOE. The spin-lattice relaxation time (T1) for all resonances used in the procedure can be measured with an inversion recovery pulse sequence. If a 90 pulse is used for excitation, quantitation at the level of 99.3% may be achieved with a recycle time, Tr (the sum of the relaxation delay and aquisition time), of 5 × T1, and improved to even higher levels by using longer recycle times or shorter pulses. The general equation for the degree of quantitation, Q, of a resonance as a function of the pulse angle, , and Tr, and T1 is given by:
+'&img=../../images/v38332/c1761-eq22-usp36.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
If a 45 excitation pulse is used, with a Tr = 5T1, Q = 0.998. However, the accuracy of quantitation in the final spectrum depends not only on the Q values of the resonances but also on the accuracy of the integration method and on the S/N ratio in the spectrum. Hence, Q values somewhat less than unity may be warranted and other angles and recycle times should be used.
The minimization of systematic quantitation bias should be sufficient for the intended use of the procedure. Alternatively, quantitative procedures may be developed using conditions for which Q is not unity for some or all resonances, provided the value of Q is precisely known and corrected for.
For quantitative methods using heteronuclei, the possibility of differential NOEs should be avoided by using a Tr at least 5 times the longest T1 value and by using inverse gated decoupling (decoupler gated on only during the acquisition time). Preferably, 90 pulses should be used. For quantitative methods, relaxation agents are often used for shortening the T1 values of heteronuclei.
The reproducibility of an NMR method depends on a variety of acquisition and processing parameters, all of which should be described in the procedure. These include pulse angle, acquisition time, relaxation delay, spectral width, number of points in the FID, number of acquisitions, number of points used (if any) for zero filling, line broadening, baseline correction, integral breaks, and temperature. For best reproducibility, integral breaks should be specified to 0.01 ppm for 1H NMR methods and to 0.1 ppm for 13C methods.
Quantitative analysis, as well as detection of trace impurities, has markedly improved with modern instrumentation. Stronger magnetic fields and improved probe technology have enhanced the sensitivity of NMR procedures in recent years.
SOLID-STATE NMR
The analytical usefulness of solid-state NMR spectroscopy for studying solid materials lies in the fact that the same types of nuclei in different solid-state environments exhibit different resonance frequencies. Applications of this technique in pharmaceutical analysis range from solid form (polymorph, solvate) identification and quantitation in bulk drug substances to physical/chemical profiling of dosage forms. The technique has the unique ability to probe electronic environments of specific nuclei in the solid state over a large timescale without the requirement of single-crystal substrates or even homogeneous samples. Methods and procedures presented herein are directed at observing 13C, the most popular NMR nucleus for solids. The concepts may be equally applied to other relevant spin-1/2 nuclei such as low-natural-abundance 15N as well as high-natural-abundance 31P.
Cross-Polarization Magic Angle Spinning (CPMAS) NMR Technique
The basic principles of NMR are the same for solution and solid-state measurements, but conventional solution-phase 1-D NMR data acquisition techniques do not normally produce detectable spectra for solid samples because of low sensitivity and extensive line broadening. The sensitivity of the solid-state experiment is low for 13C based on its 1.1% natural abundance and long spin-lattice (T1) relaxation times. Line broadening arises primarily from dipole–dipole interactions and chemical shift anisotropy (CSA), which are not averaged to zero because of the fixed orientation of molecules in a packed solid sample vs the rapid molecular tumbling of the molecules when they are in solution. If not averaged, CSA results in the simultaneous observation of all different orientations of molecules with respect to the applied magnetic field. CSA patterns may span the width of an entire liquid spectrum. Three modifications to standard solution methods—cross-polarization (CP), magic angle spinning (MAS), and high-power 1H decoupling—are routinely used in combination to obtain high-resolution solid-state NMR spectra.
cross-polarization (CP)
CP addresses the low sensitivity associated with collecting NMR spectra of dilute spin-1/2 nuclei such as 13C. CP is a double-resonance procedure wherein abundant 1H and rare 13C spins are brought into resonance by simultaneously applying two spin–locking rf fields (B1H and B1C), the magnitude of which will satisfy the Hartmann–Hahn matching condition,
During this contact time, polarization transfer occurs allowing the rare 13C spins to take on the magnetization and relaxation behavior of the abundant 1H spins, leading to a sensitivity enhancement (up to four-fold based on the ratio of the 1H and 13C magnetogyric ratios) and a reduction in the pulse repetition time. Reducing the pulse repetition time allows a greater number of aquisitions to be accumulated per unit time, which yields a better S/N ratio. In instances where it may be difficult or impossible to record CP spectra because of weak 13C–1H coupling or short spin-lattice relaxation times in the rotating frame (T1rH), direct polarization (Bloch decay) may be the only approach to recording solid-state NMR spectra.
magic angle spinning (MAS)
Line broadening in solid-state NMR is eliminated or averaged by both MAS and high-power 1H decoupling. MAS involves mechanically rotating the sample at an angle of 54.7 (the magic angle) relative to the static magnetic field in order to simulate rapid molecular tumbling in solution. Rotating a solid sample at the magic angle to minimize line broadening requires that the sample be spun faster (in Hz) than the width of the CSA. High spinning rates are possible with current MAS probe technology, but complications can arise with CP and may require techniques such as ramped-amplitude CP (RAMP-CP) and variable-amplitude CP (VACP) to improve the efficiency of magnetization transfer from 1H. Additionally, MAS can raise the sample temperature significantly if it is not controlled, and pressures at the periphery of the rotor may be thousands of times the ambient pressure. These stresses may induce phase transformations, loss of solvent, and other effects.
sideband suppression
As shown in Figure 9, slower spinning rates can be used to avoid compromising the solid sample, but when CSA is incompletely averaged, spinning sidebands will appear in solid-state NMR spectra. These artifacts are separated from the centerbands by integer multiples of the spinning rate (in Hz) and can be readily identified as the peaks that shift in spectra acquired at different spinning speeds. Spinning sideband manifolds contain useful information but can interfere with the signals of interest and may be particularly problematic when one uses higher field instruments. The total suppression of spinning sidebands (TOSS) procedure is commonly used to eliminate spinning sidebands from solid-state NMR spectra.
+'&img=../../images/v38332/c1761_fig9.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 9. 13C CPMAS NMR spectra of polycrystalline -glycine collected at different sample spinning (MAS) rates. Spinning sidebands appear in solid-state NMR spectra when the sample spinning frequency is less than the width of the CSA pattern.
high-power 1h decoupling
High-power 1H decoupling is used to further reduce the line broadening from dipolar coupling to 1H spins in the solid state. Specialized hardware is required to deliver the rf power needed for 1H decoupling in solids, an rf power that is two orders of magnitude greater than that required to remove scalar coupling in liquids. CW decoupling is commonly used in solid-state NMR, although two-pulse phase-modulated and small phase incremental alteration (SPINAL-64) decoupling are increasingly used to improve the sensitivity and resolution of dilute spin spectra.
Typically only 1H–13C heteronuclear dipolar interactions are significant when one acquires solid-state 13C NMR spectra of organic materials. Homonuclear 13C–13C dipole–dipole and scalar coupling interactions are negligible. Because of their low natural abundance, the probability that two 13C nuclei are in close proximity is very small. However, homonuclear 13C–13C dipolar coupling can be a concern for 13C-labeled substrates.
experimental setup
The basic setup for a CPMAS experiment will necessarily include magic angle setting, shimming, pulse calibration, Hartmann–Hahn matching, and spectral referencing, each of which is typically conducted on standard samples. Accurate measurement of the pulse lengths and associated rf power levels is essential for solid-state NMR experiments. Setting up the magic angle, shimming a CPMAS probe, and measuring its sensitivity for different tuning ranges are parts of probe setup and performance assessment. Shimming a solid-state NMR probe for MAS is more complicated than that for a solution probe because shim gradients are designed for the vertical orientation of solution NMR tubes. Solid-state NMR probes have no 2H lock channel, so shimming must be performed manually. Because of the intrinsically broad peaks, shimming is not nearly as critical in solid-state NMR as it is in solution NMR.
Standard compounds used for setting the magic angle and optimizing pulse lengths and the associated rf power levels for CP are listed in Table 6. KBr is commonly used for magic angle adjustments, observing the 79Br resonance and adjusting the sample spinning angle to maximize (to 10 ms and beyond) the duration of the rotational echo train in the FID. Liquid samples can be used to shim solid-state NMR probes, although solid adamantane is commonly used for this purpose. Adamantane, glycine, and hexamethylbenzene (HMB) are commonly used for Hartmann–Hahn matching and testing sensitivity. Typically, the reference sample for testing sensitivity is permanently packed into a rotor to ensure that the same amount of sample is used.
Table 6. Standard Samples Commonly Used for Setting Up CPMAS Experiments
| Setup Procedure | Nucleus | Standard Sample(s) |
|---|---|---|
| Setting the magic angle | 79Br | KBr |
| Shimming | 13C | adamantane |
| Pulse calibration | 1H, 13C | adamantane, HMB |
| Hartmann-Hahn matching | 1H/13C | adamantane, HMB |
| Sensitivity | 13C | HMB, adamantane, |
Solid-state NMR spectrometers are generally used without field/frequency locking, so the resulting chemical shifts are less accurate than those for solutions. Calibration of the chemical shift can be done using either a primary or secondary standard. Spectral referencing is typically performed by sample replacement (external referencing). Standard compounds commonly used for spectral referencing in solid-state NMR are listed in Table 7. Note that glycine is polymorphic, so its crystal form should be ensured before its use in referencing spectra.
Table 7. Reference Compounds Commonly Used for Solid-State NMR
| Nucleus | Primary Standard | Secondary Standard(s) | Chemical Shift in ppm from Primary Standard |
|---|---|---|---|
| 13C | TMS | HMB adamantane |
17.35 (CH3) 38.48 (CH2) 176.45 (carboxyl) |
| 15N | nitromethane | NH415NO3
15NH4Cl |
|
| 31P | 85% H3PO4 | CaHPO4 · 2H2O (brushite) | 1.4 |
| 29Si | TMS | tetratrimethylsilylmethane | |
| 19F | CF3Cl | perfluorobenzene |
General Test Procedure
Spectrometer performance should be demonstrated first for a reference sample as described in Cross-Polarization Magic Angle Spinning (CPMAS) NMR Technique, Experimental Setup. The magic angle, pulse lengths, and associated rf power levels for CP that are established using the reference compounds are sample independent. To obtain a CP spectrum of the sample, only the recycle delay needs to be chosen, followed by the contact time. When quantitative signal intensities are not required, an optimum recycle delay, i.e., one that affords the best S/N ratio, is 1.2T1H, and the contact time is generally that which provides the optimum S/N ratio or that which best shows the features of most interest. For quantitative CP, a recycle delay of at least five times the longest T1H of a heterogeneous mixture is suggested to ensure full relaxation, and a full analysis of the CP signal as a function of contact time must be conducted. See the subsection Quantitative Analysis.
sample preparation
Sample-handling procedures used in solid-state NMR are substantially different from those used in liquid NMR. Solid samples are packed in ceramic rotors that are capped with fluted drive tips specifically designed for MAS. Fine powders are typically tamped into MAS rotors, although solid plugs, e.g., whole tablets, can be cut to fit the exact inner dimensions of the rotor and can be inserted directly into the rotor. Crushing or grinding may be used to reduce the sample to a fine powder, but caution should be used in order not to induce phase transformations. Depending on the compressibility of the powder and the rotor volume, 40–400 mg of material is typically required to fill a sample rotor.
Physical Characterization
Specific components in heterogeneous systems can be searched based on unique nuclei or different NMR relaxation properties. The identification of crystalline and amorphous materials can be accomplished by comparison of the solid-state NMR spectrum of the sample preparation with that of a reference standard. Chemical shifts and relative peak intensities can be used in the comparison. Amorphous materials generally give good MAS spectra with broader peaks than those seen for crystalline materials. Highly crystalline samples typically give 13C linewidths of the order of a few tens of Hz. The shape of the signals generally is between Lorentzian and Gaussian, a fact that should be recognized in deconvoluting overlapped spectra.
Relaxation
Relaxation parameters of interest in solids include spin-lattice relaxation (T1), spin-lattice relaxation in the rotating frame (T1), spin-spin relaxation (T2), and cross-relaxation (TCP). In organic solids, 1H spin diffusion is generally efficient so that pure compounds normally give single values for each of the relaxation times, T1 and T1. For CP experiments, T1H is used to establish the recycle delay between acquisitions. T1H relaxation times can be measured using either progressive saturation or inversion recovery pulse sequences. In CP experiments, TCP and T1H characterize the magnetization build-up and decay, respectively. T1H is measured via the 13C signal using a delayed contact CP pulse sequence that has a variable delay time before the CP contact.
Quantitative Analysis
To quantitatively assess solid-state NMR spectra under CP conditions, extra measures must be taken. In addition to ensuring that the sample spinning axis is precisely set to the magic angle (54.7) to minimize CSA broadening, the temperature and spin rate must be carefully controlled and the MAS probe properly tuned. Suggested recycle delays of 5T1H are allotted between successive pulses to ensure that the magnetization has returned to its full equilibrium value. Both TCP and T1H relaxation must then be accounted for in the selection of the contact time. Typically, the CP contact time chosen is that which provides maximum sensitivity for the signals of interest. Quantitative analysis can be performed using either internal or external referencing methods. The use of internal standards can compensate for variability in sample volume and B1 inhomogeneity throughout the sample.
By properly setting data acquisition parameters (recycle time, pulse widths, contact time, Hartmann–Hahn match, and decoupling power for each chemical system), signals can be obtained that are proportional to the number of nuclei producing them. For quantitative analysis, integrated signal intensities should be used rather than peak heights because linewidths in solid-state spectra often vary. When spinning sidebands are not eliminated by MAS, the intensity of the spinning sideband manifold must be added to the centerband intensity.
Spectral Editing
A key step in the analysis of any NMR spectrum is the assignment of individual resonances to unique phases and, in some cases, to specific atoms in the molecule. Special pulse sequences are available and may assist in simplifying CPMAS spectra and assigning signals. Dipolar dephasing, also known as nonquaternary suppression or interrupted decoupling, yields spectra that typically contain signals only from quaternary and methyl carbons. Spectral subtraction of dipolar dephasing spectra from normal CP spectra or short contact time CP may be used to produce subspectra that contain signals from methylene and methine carbons only. Polarization-inversion techniques can be used to identify methylene and methine carbons.
LOW-FIELD NMR
Low field NMR (LF-NMR), sometimes referred to as time domain NMR (TD-NMR), experiments are performed by measuring relaxation, relaxometry, or diffusion. Instrumentation for these applications is based on low-field permanent magnet technologies that operate in the 2–25 MHz frequency range. Inexpensive stationary bench-top and portable TD-NMR spectrometers are commercially available. Typical bore sizes are 10–50 mm in diameter. A recent development in spectrometer design uses a mobile mouse probe as an alternative to a stationary magnet. This design allows analysis of samples of unrestricted size.
Most TD-NMR applications are based on simple pulsing sequences, including FID, Hahn–echo, Carr–Purcell–Meiboom–Gill, and solid–echo acquisition. The choice of pulse sequence depends on the physical and chemical properties of the sample as well as the information that is desired from the experiment. These systems can be used to measure longitudinal (spin-lattice, T1) and transverse (spin-spin, T2) relaxation times. Diffusion properties of compounds can be exploited using pulsed field gradient (PFG-NMR) experiments.
The classical application of relaxometry is for the determination of food product components based on differences in longitudinal and transverse relaxation times of water, fats, and proteins.
Relaxivity
The magnitude of a substance’s capacity to enhance the relaxation rate of a nucleus is referred to as relaxivity, expressed in units of sec1mM1. An example of such a substance commonly used in NMR spectroscopy is paramagnetic chromium acetylacetonate. Paramagnetic species are used in the medical industry as contrasting agents for magnetic resonance imaging. The relaxivity of a substance is determined experimentally by measuring the spin-lattice relaxation time (T1) of a test substance and plotting 1/T1 against the concentration in units of mM (mmol/L). The slope of the curve is the numerical relaxivity.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Kahkashan Zaidi, Ph.D.
Senior Scientific Liaison (301) 816-8269 |
(GCCA2010) General Chapters - Chemical Analysis |
USP38–NF33 Page 1659
Pharmacopeial Forum: Volume No. 37(6)