



INTRODUCTION
Immunological Test Methods (ITM) utilize binding between an antigen (Ag) and antibody (Ab). (See Appendix 1 for a complete list of acronyms used in this chapter.) Enzyme-linked immunosorbent assay (ELISA) is one of the most widely used ITM for characterization, release, and stability testing of biotechnology products to help ensure the quality of biological drug substances and drug products. The term ELISA is used here in a broader sense and includes enzyme immunoassays (EIA), as well as alternative detection methods, e.g., chemiluminescence and fluorescence.
This chapter provides analysts with general information about principles, procedures, experimental configurations, assay development, and validation for solid-phase ITM like ELISA and can be used for the other immunoassay variations mentioned above. The chapter also covers reference standard(s) and control(s) used for immunoassays. The information can be adapted to the specific procedures of a monograph. This chapter does not cover immunoassays for the measurement of immune responses to product in animals or humans (e.g, serological or cellular assays), non-immunoassays (e.g, receptor-ligand interactions), or other related approaches.
The chapter is part of a group of general information chapters for immunological test methods [Immunological Test Methods—General Considerations  1102
1102 , Immunological Test Methods—Immunoblot Analysis 1104 (proposed), and Immunological Test Methods—Surface Plasmon Resonance 1105], and also is related to the general information chapters for bioassays [Design and Development of Biological Assays 1032, Biological Assay Validation 1033, and Analysis of Biological Assays 1034].
, Immunological Test Methods—Immunoblot Analysis 1104 (proposed), and Immunological Test Methods—Surface Plasmon Resonance 1105], and also is related to the general information chapters for bioassays [Design and Development of Biological Assays 1032, Biological Assay Validation 1033, and Analysis of Biological Assays 1034].
Definition
ELISA can be defined as a qualitative or quantitative solid-phase immunological method to measure an analyte following its binding to an immunosorbent surface and its subsequent detection by the use of enzymatic hydrolysis of a reporter substrate, either directly (as with an analyte that has enzymatic properties or is directly labeled with an enzyme) or indirectly (by means of an enzyme-linked antibody that binds to the immunosorbed analyte). Qualitative results provide a simple positive or negative result for a sample. Converting quantitative to qualitative results based on a cutoff value that separates positive and negative results is common practice. Because the performance properties of the assay depend heavily on the cutoff value, the process used to determine the cutoff should be evidence-based and well documented. Quantitative assays determine the quantity of the analyte based on the interpolation of a standard calibration curve with known analyte concentration, run simultaneously in the same assay. This standard should be an appropriate, preferably homologous, reference or calibration material that is representative of the analyte(s) of interest. The power of immunoassays has been demonstrated by the variety of procedures that have evolved, including alternative solid surfaces such as beads of different sorts, various plastics in plates of different configurations, and alternative detection methods, e.g., chemiluminescence and fluorescence. ELISA assays are widely used in the biopharmaceutical industry for various applications such as identity, purity, potency, detection or quantitation of antibody or antigen, and other purposes.
Basic Principles
The essential steps of an ELISA can be broken down as follows (see Figure 1):
+'&img=../../images/v38332/c1103-f1-pf375.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 1. Essential steps for performing an ELISA.1
1. Binding of the capture reagent (generally an antibody or antigen), which functions as an immunosorbent for capture of the analyte, to a solid surface;
2. Removal of excess, unbound capture reagent followed by blocking of unoccupied binding sites with a blocking protein such as albumin, gelatin, casein, or other suitable material;
3a. Incubation of the analyte (in the test sample or reference standard) with the capture reagent to bind the analyte onto the solid surface, followed by the washing away of unbound material in the test sample and detection of the analyte. Direct detection occurs when the analyte has enzymatic activity or has been linked to a detector molecule (e.g., enzyme); or
3b. Incubation of the analyte (in the test sample or reference standard) with the capture reagent to bind the analyte onto the solid surface, followed by the washing away of unbound material in the test sample and subsequent detection of the analyte (Figure 1, step 3a). Indirect detection occurs when the analyte is detected by the addition of a secondary enzyme-labeled reagent (Figure 1, step 3b); and
4. Quantification of the analyte by addition of a substrate suitable for the detector used (e.g., TMB, 3,3¢,5,5¢-tetramethylbenzidine), followed by comparison of the test sample to the reference standard.
ASSAY DESIGN
Five general categories of ELISA are described in Table 1 and in the sections that follow. The assay designs are flexible and, depending on specific needs, can be modified from these procedures. The choice of format depends primarily on the amounts and purity of reagents and equipment available. On some occasions the analyte being characterized actually is an antibody, as in the case of a monoclonal antibody that is being developed as a drug. In this case, anti-idiotypic or other antibodies specific for the antibody are used to develop the assays.
Table 1. Representative ELISA Types
| ELISA Type | Required Reagents | Attributes | Disadvantages |
|---|---|---|---|
|
Direct detection |
|
|
|
|
Indirect detection |
|
|
|
|
Competitive |
|
|
|
|
Sandwich |
|
|
|
|
a
This reagent can be either purified or partially purified. The terms “analyte” and “antigen” are used interchangeably when describing ELISAs.
|
|||
Direct ELISA
Directly Labeled Antibody:
In this assay an antigen is coated onto a solid surface and the remaining unbound reactive sites are blocked (Figure 2A). Then a solution containing a specific antibody labeled with a detector is added. After incubation, the unbound antibody is washed away, followed by the addition of an appropriate substrate for the detector used.
+'&img=../../images/v38332/c1103-f2-pf375.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 2. Schematic representations of direct, indirect, competitive, sandwich, and bridging ELISAs.2 (Ab = antibody; Ag = antigen (or analyte); Preincub = preincubation)
Directly Labeled Antigen:
This assay is similar to that using a directly labeled antibody except that the antibody is coated onto the solid surface and a labeled antigen is used as the detector.
Indirect ELISA
In this assay an antigen is coated onto a solid surface and then, after blocking, a solution containing a specific antibody is added (Figure 2B). After incubation, the unbound antibody is washed away, followed by the addition of an anti-immunoglobulin (anti-Ig) detector antibody. Anti-Ig detectors are available commercially for specific Ig classes and subclasses from a variety of species, which makes this assay format useful for isotyping of antibodies. In addition, the use of a labeled anti-Ig detector amplifies the signal compared to a Direct ELISA, thereby increasing assay sensitivity.
Competitive ELISAs
Direct Antibody Competitive ELISA:
This assay is used to detect or quantitate soluble antigens (Figure 2C). It requires an antigen-specific antibody that has been conjugated to an appropriate detector, e.g., horseradish peroxidase, alkaline phosphatase, ruthenium, or fluorescein. It also requires a purified or partially purified antigen for coating. The antigen is coated onto a solid surface, followed by a blocking step. The antibody–conjugate is incubated with the test solution containing soluble antigen. The mixture is then added to the immobilized antigen, incubated, and unbound antigen-antibody complex is washed away. Substrate is added, and the inhibition of the reaction (e.g., colorimetric, electrochemiluminescence, fluorescence, or chemiluminescence) is measured relative to the reaction when no competitor antigen is added. The amount of inhibition is inversely proportional to the amount of antigen in the test sample. Competitive assays can also measure small molecules by coating an antibody to the plate that is specific to the small molecule. The small molecule is often biotinylated with a long linker that does not interfere with binding between the capture antibody on the plate and the small molecule. Antigen (the small molecule) in the sample then competes with the labeled small molecule for binding to the capture antibody. After washing, a detection reagent (e.g., streptavidin labeled with HRP) is added to detect the binding complex.
Direct Antigen Competitive ELISA:
This assay is similar to the Direct Antibody Competitive ELISA, except that it is used to detect soluble antibodies. The antigen is conjugated to the detector and the antibody is coated onto the solid surface.
Indirect Antibody Competitive ELISA:
This assay is similar to the Direct Antibody Competitive ELISA, except that instead of directly labeling the antibody, the test uses a labeled anti-Ig reagent for detection.
Indirect Antigen Competitive ELISA:
This assay is similar to the Direct Antigen Competitive ELISA, except that instead of directly labeling the antigen, the test uses a labeled secondary antibody for detection.
Sandwich ELISA
Direct Sandwich ELISA:
In this assay an antibody is immobilized onto a solid surface and blocked, and then a solution containing a specific antigen is added (Figure 2D). After an incubation step, the unbound material is washed away, and a labeled detector antibody is added. This assay format requires two antibodies, each of which binds to different epitopes on the surface of the large and complex molecule. The two antibodies are specific for the antigen, and the antigen should be sufficiently large and complex to accommodate the binding of two antibodies.
Indirect Sandwich ELISA:
Alternatively, instead of directly labeling the detector antibody, an anti-Ig antibody detector can be used. Indirect sandwich immunoassay formats can be considered only if each binding reagent is from a unique species (e.g., a sandwich assay using two mouse monoclonal antibodies for capture and detector could not be detected indirectly because the resulting signal may become independent of the antigen concentration).
Bridging ELISA:
This subset of Sandwich ELISA assays often uses a single antibody for both capture and detection (Figure 2E). If a monoclonal antibody is used, it requires that the target antigen have at least two identical epitopes that are adequately spaced to prevent steric hindrance so that one epitope binds to the capture antibody and the other epitope binds to the detector antibody. Alternatively, a polyclonal antibody can be used but still requires that the target antigen be large enough to accommodate the binding of two antibody molecules. With respect to specificity and sensitivity, bridging assays usually are suitable for most large molecules.
CHOICE OF ASSAY
Deciding which ELISA procedure or format to use often depends on individual choice and availability of reagents, instruments, and other equipment. For example, sometimes a laboratory repeatedly engineers a particular epitope into multiple fusion proteins. In this case, the laboratory can use certain common qualified reagents (e.g, an antibody to a glutathione S-transferase region in multiple fusion proteins), facilitating rapid sandwich immunoassay development. Small antigens with a limited number of epitopes available for antibody binding restrict ELISA format choices. If there is only one binding epitope, then ELISA methods that use the sandwich/two-site binding or other bridging formats cannot be used because they require at least two available epitopes for antibody binding. In addition, small molecules are not usually used as a capture reagent on a plate because the process may interfere with binding to the detection reagent. Examples of such small molecules are some peptides, oligosaccharides, nucleotides, and antibacterials. Analysts usually adopt a competitive assay format for such small analytes.
Different assays and formats may demonstrate different properties and characteristics, e.g., specificity, precision, accuracy, sensitivity, dynamic range, dose-response ratio, sample throughput, sensitivity to interference, and simplicity or efficiency for automation. Ease of validation also may vary between different assay protocols and formats. Assay designs with replicates in adjacent wells could be biased if there are location effects; hence, in this case, replicates should not be in adjacent wells. Assay designs that are convenient to perform on 96-well plates, using relatively few single-channel pipet actions and more multi-channel pipet actions, are usually easier to adapt to automation. Assays with steep dose-response curves are generally better able to deliver high precision estimates; however, some assays with steep dose-response curves are imprecise in the EC50 and require a wider dose range.
PROCEDURES
Solid Phase
Solid phases are available in a variety of forms (e.g, membrane, plate, or bead) and chemistries (e.g, nylon, nitrocellulose, polyvinylidine fluoride (PVDF), polyvinyl, polystyrene, or a chemically derivatized surface). The selection of the solid phase determines the most likely binding mechanism, i.e., hydrophobic, hydrophilic, or covalent interactions. In general, compared to plates, beads offer higher capacity and are more commonly used in clinical assays whereas plates are more commonly used to test biotechnology products. Additional information on plates is provided below.
Coating the Solid Phase—Immobilization of Capture Reagent:
Capture reagents are coated onto a solid phase by adding a solution containing the capture reagent to the surface. The most commonly used solid-phase materials for capture reagent immobilization are plastic 96-well microtiter plates. Those with flat-bottom wells are recommended for spectrophotometric readings, and round-bottom well plates are useful for visual assessment of a dye’s color development. The degree of coating is influenced by the concentration of capture reagent, temperature during coating, duration of capture reagent adsorption, the surface properties of the solid-phase material, and the nature of the buffer of the capture reagent solution. Although the optimum coating concentration must be determined for each capture reagent, concentrations of 1–10 µg/well are most commonly used. The volume of capture reagent added to each well usually corresponds to the sample volume that will be analyzed, i.e., 50–100 µL. Coating duration, temperature, and buffers are discussed separately below. During the coating procedure analysts should avoid introducing bubbles. Proteins that bind to plastic can be denatured, which alters antigenicity. In such cases, a capture antibody or an intermediary protein such as Protein A or Protein G can be used. In addition, streptavidin can be used if the reagent is biotinylated. The pH of the coating buffer should be optimized based on the isoelectric point of the capture reagent and the surface properties of the assay plate chosen.
Microtiter Plates:
The composition and commercial source of the microtiter plate can influence binding of the capture reagent during coating. Several microtiter plates from different suppliers should be compared using a single coating procedure to select those that provide high specificity for the capture reagent of interest and low nonspecific background. Comparisons of different grades of plates from a single supplier also may be needed. Clear plates typically are used for colorimetric ELISA, and opaque plates often are used for chemiluminescent and fluorometric ELISA. Acidic capture reagents may require a lower pH solution to neutralize repulsive forces between the protein and solid phase. Peptides often require optimization of buffer pH based on their charge for optimal coating conditions during assay development. Polysaccharides, lipopolysaccharides, or glycoproteins may be difficult to coat directly to the plate and may require a capture antibody or a buffer that contains lysine or glutaraldehyde. Coating with an antibody can be enhanced by precoating the microtiter plate with Protein A or Protein G or a combination of the two, which allows binding to the Fc region so that the Fab portion can bind to the analyte of interest. However, care must be taken to ensure that subsequent secondary antibodies do not react with the Protein A- or Protein G-coated wells. In this case, for example, chicken IgY or another appropriate antibody class could be used. Microtiter plate formats other than the 96-well variety, such as half volume 96-well or 384-well plates, can be used to increase throughput and/or conserve reagents.
Coating Time:
Coating time depends on binding kinetics, stability, concentration of capture reagent, and incubation temperature. Although different combinations of coating times and temperatures often result in the same coating efficiency, the stability of the capture reagent (which should be determined during method development) influences which conditions to select. Analysts must assess the impact of varying the coating time in order to determine the robustness of the assay procedure.
Coating Temperature:
Coating temperature and time are closely related assay parameters. The coating temperature depends on the binding kinetics and stability of the antigen. Higher temperatures can increase the rate of adsorption and may shorten the coating time, but they are likely to affect interaction sites and to reduce antigen-antibody affinity. Typical combinations of time and temperature are 1–4 h at ambient temperature, 15 min to 2 h at 37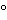 , or overnight at 4. Analysts should determine the effects of variations in temperature in order to assess the robustness of the assay procedure.
, or overnight at 4. Analysts should determine the effects of variations in temperature in order to assess the robustness of the assay procedure.
Buffers:
Buffers used for diluents, coating, blocking, and washing plates can affect overall assay performance. Buffer components can interact with the test sample and inhibit binding. They also can cause low antigen sensitivity or high nonspecific background activity.
Diluent—
Buffers [e.g., phosphate-buffered saline (PBS) or imidazole-buffered saline] with polysorbate 20 (0.01%–0.1%) are used commonly for different ELISA steps as a diluent and washing buffer.
Coating Buffers—
Coating buffers should maximize assay consistency and promote binding of the capture reagent to the solid phase. Commonly used coating buffers include 50 mM carbonate, pH 9.6; 20 mM Tris-HCl, pH 8.5; and 10 mM PBS, pH 7.2. The choice of coating buffer depends on the nature of the individual antigens and should be determined empirically.
Blocking Agents and Buffers—
A blocking agent is a compound (e.g., protein or detergent) that should saturate the remaining immunosorbent binding sites following capture reagent (antibody or antigen) binding. This reduces nonspecific binding of analyte and nonanalyte components to the immunosorbent matrix and/or the absorbed reagent. Nonspecific binding occurs when protein in the test sample binds to the plastic of the microtiter plate or absorbed reagent instead of specifically binding to the capture reagent of interest. Nonspecific binding can be reduced by adding blocking reagent to the wells and by the addition of another protein such as bovine serum albumin (BSA) to the dilution buffer. The choice of blocking agent should be governed by the nature of the capture reagent, plate, coating buffer, test sample diluent, and related factors. If any of these parameters changes, a change in blocking agent may be needed. Commonly used blocking agents include BSA, nonfat milk, gelatin, casein, normal horse serum, fetal bovine serum, polysorbate 20, and others. Several grades of BSA are available commercially, and the optimal grade should be empirically determined for each assay. In addition, many commercial blocking and assay diluent reagents are available for ITMs.
Adding Samples and Reagents
Samples and reagents generally are pipetted into the ELISA plate wells. Care should be taken to avoid cross-contamination, frothing, or bubbles. Labor-saving equipment such as electronic pipets, automated liquid handlers, plate washers, and robotic pipets also can be used to improve precision, reduce analyst-to-analyst variability, and increase throughput.
Pipets:
Single, multichannel, and robotic pipets with set or fixed volumes are available. The type and accuracy of pipets should be evaluated for each application. Regular maintenance and professional calibration of pipets should be performed and documented.
Pipet Tips:
A variety of pipet tips are available, some of which are specific to the type of pipet. The type and accuracy of the pipet tip, particularly related to the viscosity and nonspecific binding of the materials, should be evaluated for each application.
Washing
Wash steps are included throughout the ELISA procedure to remove the unbound coating antigen, sample, and detection reagents. Washing is critical for assay performance, can be a source of assay failure, and is important to evaluate during method development. Multiple approaches can be used for washing. Manual procedures include using a squeeze bottle, dipping the microtiter plate in wash buffer, and adding wash buffer with a multichannel pipet or hand-held multi-channel (8- or 12-pin) manifolds. Analysts should wash carefully to avoid cross-well contamination. Automatic microplate washers generally provide more washing consistency. Strip-well and multiwell washers are available. Most automatic washers can be programmed for different dispensing volumes and speeds, number of washes, speed of buffer aspiration, and amount of residual buffer left in the well. Incorrectly programmed or maintained as well as incompletely cleaned automatic washers can cause assay variation and elevated assay background.
Incubation
ELISAs are incubated following the addition of samples and reagents. The optimal time, conditions, and temperature of each incubation step should be determined during method development. Incubation times vary from minutes to overnight. Commonly used incubation temperatures are ambient temperature, 4, and 37. ELISA plates commonly are sealed or placed in a secondary container to avoid evaporation or contamination during incubation. Atmospheric conditions such as dry or humidified incubation should be evaluated during method development. Rocking, shaking, or rotating the microtiter plates may be necessary or desirable depending on the kinetics of binding.
Blocking Conditions and Nonspecific Reactions
After immobilization and removal of the unbound antigen or antibody, unoccupied binding sites are blocked to ensure that the measured analyte in the test article or subsequent (detection) reagents does not bind nonspecifically to the solid surface or to the coated antigen or antibody. If nonspecific binding occurs, any reported signal could bias the measurement and may reduce the sensitivity and dynamic range of the assay. Blocking is critical to ensure the sensitivity and/or specificity of the assay. Sources of nonspecific binding fall into two general categories:
- Ionic or hydrophobic interactions occur when binding is mediated by nonspecific ionic or hydrophobic interactions between assay reagents and the solid surface or another assay reagent.
- Immunological interactions occur when binding is mediated by unintended antigen-antibody interactions. This occurs when antibody preparations used in the assay interact with other assay reagents. For example, if an ELISA was designed to test a serum-derived analyte using murine capture and detection antibodies, antibodies in the test article with reactivity to murine Ig (also known as heterophilic antibodies) could be nonspecifically detected in the assay.
The choice of blocking agent (examples are found in the Blocking Agents and Buffers section above) is determined empirically, and the balance between the reduction in nonspecific binding and the impact on assay sensitivity should be assessed during method development. Cross-reactivity with other assay reagents should be considered; for example, endogenous biotin is found in milk and serum, and serum may contain antibody to viral or bacterial proteins. Therefore, screening of serum lots may be necessary. The volume of blocking solution added to the well should be greater than the maximum reaction volume used for later steps so that all of the potential surface area that may interfere with the binding reaction is blocked.
In addition, Ig in the test materials can be removed by using buffers that inhibit antibody conformation or aggregate the heterophilic antibodies, by blocking with nonimmune serum, or by removing Fc regions in critical antibody reagents, thereby reducing or eliminating undesired immunological interactions that cannot be addressed by the blocking reagents described above. Negative control wells can be included to monitor nonspecific reactions. The nature of the negative control wells depends on the assay but can include blocked wells without coating antigen, eliminating the primary or secondary antibody, or using buffer in place of sample. Control wells also can be useful as part of system suitability testing.
Pretreatment of Samples
Although ELISA methods are designed to measure an analyte in complex mixtures, the presence of other materials can prove problematic if they interfere with analyte detection. In order to ensure assay specificity, the specific procedure to treat samples to remove nonspecific interfering substances (e.g., reducing agents or precipitates) can be determined empirically during method development and then can be incorporated into the validated assay. Any sample-processing step should be evaluated against the potential that the treatment will alter the test article’s properties and/or introduce further variability that results in biased measurements. Samples, standards, and controls should be prepared and handled in processes as similar to each other as possible. Analysts should verify that sample pretreatments have not damaged the sample so much that it can no longer be measured (e.g., by spiking experiments).
Detector Antibodies
Depending on ELISA format, detector antibodies labeled with enzyme or other labels can be used as primary or secondary reagents to enable detection of the immobilized analyte. In a direct or competitive ELISA (Figure 2A and Figure 2C), after the analyte is bound to the immunosorbent surface, excess analyte is washed away and the immobilized analyte is detected using a detector antibody that is considered to be the primary antibody. In other ELISA formats (Figure 2B, 2D, and 2E), the analyte-specific Ig (nonconjugated primary antibody) is allowed to bind to the immobilized analyte, and any excess antibody is washed away before the addition of a detector antibody, which is termed the secondary antibody.
To facilitate detection, in all ELISA formats that use enzyme-conjugated antibodies, a substrate specific for the conjugated enzyme is introduced into the assay system. An enzymatic reaction ensues, converting a substrate into a soluble product that can be measured using appropriate wavelengths and a suitable reader.
ELISA sensitivity depends on the quality of the reagents and the detection system, including the label and substrate. If multiple differently conjugated antibodies are available, analysts should select one appropriate for the assay. During this evaluation, the dilution of each conjugate that yields desirable sensitivity and specificity should be determined using appropriate controls.
The most commonly used labeling enzymes for conjugating to antibodies include alkaline phosphatase (AP), horseradish peroxidase (HRP), and galactosidase. These enzymes are highly specific, sensitive, and stable in catalyzing chromogenic, luminescent, or fluorescent reactions. para-Nitrophenyl phosphate (pNPP) is a commonly used substrate for AP. Commonly used substrates for HRP include TMB, OPD (o-phenylenediamine dihydrochloride), and ABTS [2,2¢-azino-bis(3-ethyl-benzothiazoline-6-sulfonic acid) diammonium salt] (see Table 2). The substrates for AP and HRP are chromogenic and result in the formation of a colorimetric product that can be measured using a spectrophotometer. Chemiluminescent and fluorescent substrates for AP and HRP also are available, and in many cases they are available as commercial kits. Disodium 3-(4-methoxyspiro{1,2-dioxetane-3,2¢-(5¢-chloro)tricyclo[3.3.1.13,7]decan}-4-yl)phenyl phosphate (CSPD) is a known chemiluminescent substrate for AP (see Table 2). Well-known fluorescent substrates for galactosidase include MG (4-methylumbelliferyl galactoside) and NG (nitrophenyl galactoside). If a chemiluminescent substrate is used, then a luminometer is required to quantitate the formed product. A fluorometer is needed if a fluorescent substrate is used in the ELISA.
Table 2 also provides a summary of the advantages and disadvantages of different types of ELISA substrates. Colorimetric substrates have been prevalent since the origin of ELISAs and may yield robust assays that generally are more cost efficient than assays that use chemiluminescent and fluorescent substrates. Nevertheless, chemiluminescent and fluorescent ELISA methods may yield more rapid and sensitive assays with a wider dynamic range than assays that use a colorimetric readout. The final choice of readout should be governed by the assay’s purpose and the requirements of the assay.
Table 2. Enzyme Conjugates and Substrates
| Readout | Principle of the Enzymatic Reaction |
Enzyme | Substrate | Reader | Advantages | Disadvantages |
| Colorimetric | Produces a colored product that yields absorbance values directly proportional to analyte concentration | AP, HRP | pNP P TMB OPD ABTS |
Spectrophotometer |
|
|
| Chemiluminescent | Produces a light emission that is directly proportional to analyte concentration | AP | CSP D |
Luminometer |
|
|
| Fluorescent | Produces excitation-induced light emission that is directly proportional to analyte concentration | Galactosidase | MG NG |
Fluorometer |
|
|
ASSAY DEVELOPMENT AND VALIDATION PLAN
Critical Reagent Development
Key considerations for critical reagents are source, purity, specificity, and stability. For quality measurements, ITMs use reference standards along with critical reagents for analyte capture and detection. Any changes of critical biological reagents should be evaluated (see, for example, guidance contained in Design and Development of Biological Assays 1032.)
Source:
The availability and quality of the starting material should be controlled so that manufacturing of the (purified) reagent can be reproducibly and consistently performed, potentially over several decades. Because critical reagents are biological molecules, sources can range from chemical synthesis (e.g., peptides) to complex biological matrices (e.g., antibodies prepared from serum, monoclonal antibody from ascites/cell culture, or fermentation/cell culture products). When appropriate for the intended use of the assay, a single lot of a critical reagent can be manufactured to establish a substantial supply and to prevent lot-to-lot variability. In other instances it may be appropriate to include in the validation multiple lots or multiple suppliers in order to demonstrate that the assay is sufficiently robust for its intended use.
Purity:
In general, the purity of critical reagents should be assessed to ensure the removal of impurities and manufacturing process residuals that can influence reagent performance and/or stability.
Specificity:
The specificity of a critical reagent refers to its ability to capture or detect only the analyte of interest. The reagent must be specific to the analyte and should show little nonspecific binding or no cross-binding to off-target molecules in complex test materials.
Stability:
The stability of critical reagents should be empirically determined to ensure assay performance over time (issues include accuracy, precision, reproducibility, and assay drift). Long-term (months to years) stability of critical reagents under required storage conditions (e.g., with defined temperatures and containers) should be determined so that appropriate expiry dating can be assigned. Short-term (minutes to days) stability (and freeze/thaw and room temperature stability for frozen critical reagents) also is required to ensure day-to-day assay accuracy, precision, and reproducibility.
Feasibility/Pilot Studies
The steps of the process by which an ELISA method is developed, validated, and used in routine sample analysis are described below:
- Generate or purchase critical reagents to measure the analyte. Determine storage conditions and stability.
- Understand the performance goals for the assay system.
- Develop the assay to the point that there is a detectable concentration response curve.
- Perform method development/robustness testing.
- Prepare the reference/calibration standard and control and assess stability.
- Establish assay procedures, appropriate controls and limits, assay and sample acceptance criteria, and instrumentation.
- Determine method performance, and qualify method for accuracy, specificity, precision, and robustness, including qualification of all applicable sample types to be analyzed.
- Validate the assay.
- Implement the method (technology transfer) in the testing laboratory, including training and qualification of analysts.
- Monitor assay performance.
During assay development, the critical parameters and reagents that are required for the assay should be assessed and set at levels that yield desired assay performance. In many instances several parameters may be evaluated, and well-designed experiments can accelerate assay development, particularly for assessing the potential interaction of several inputs.
Many ELISA procedures are product specific, and external reference/calibration standards may not be available. The preparation and stability of reference/calibration standards should be considered early in assay development.
Assay Validation
Assay validation is executed according to guidances from appropriate regulatory bodies (e.g., ICH Q2) to demonstrate that the particular test used for an analyte is appropriate for its intended use. More information about assay validation can be found in the general information chapter Validation of Compendial Procedures 1225 or in general information chapter Biological Assay Validation 1033 if the ELISA is used as a surrogate potency assay. See Appendix 2 for additional information.
DATA ANALYSIS
The analysis of ELISA data can be simple (e.g., a linear calibration with inverse regression) or complex (e.g., a nonlinear calibration curve with inverse regression). The type and rigor of data analysis depend largely on the assay system and the intended uses of the assay. For example, data reduction may estimate a concentration (e.g., ng/mL) of an unknown sample using a calibration curve. Other approaches include estimation of the half-maximal inhibitory concentration (IC50) or effective concentration (EC50), estimation of the amount of a sample that yields the same response as the EC50 (or IC50) on a standard curve, and an estimate of the relative activity of a test sample compared to a reference/calibration standard. More extensive guidance about statistical methods for potency analysis are given in general information chapters Design and Development of Biological Assays 1032 and Analysis of Biological Assays 1034.
In general, ELISA assay curves are characterized by a nonlinear relationship between the concentration of the analyte of interest and the calculated mean response. Typically, this response curve is defined by a sigmoidal relationship of response to concentration. A wide range of mathematical models can fit standard/calibration curves, and analysts should take care in the selection of an appropriate curve-fitting algorithm. In other cases, ELISA assays are used for qualitative purposes to determine whether a sample is positive or negative based on a sensitivity threshold.
Basic Statistical Analysis
Basic statistical methods are not detailed here. General information chapter Analytical Data—Interpretation and Treatment 1010 addresses important fundamentals, including data handling; computation of means, standard deviations, and standard errors; detection of and methods to address nonconstant or nonnormal variation; detection of and management of outliers; and procedures for and interpretation of statistical tests and confidence intervals. The concepts behind validation, goals, designs, analysis, and practical methods for validation are described in general information chapters Analytical Data—Interpretation and Treatment 1010, Validation of Compendial Procedures 1225, and Biological Assay Validation 1033. General test chapter Design and Analysis of Biological Assays 111 contains guidance on combining results from independent assays.
Nonlinear Statistical Analysis
Nonlinear calibration for immunoassays draws on many sources for statistical design and analysis. These include methods for assessing and addressing nonconstant variance, designs and analysis methods for experiments with complex structures, and validation. The concepts behind linear calibration design, analysis, and inverse regression apply in nonlinear calibration, and professional statisticians can help apply these appropriately.
Reporting Results
Reported estimates of concentration should be understood as having an associated confidence interval based on the results of the validation. The reported value or estimate used to describe a sample can be based on a combined result from multiple assays.
APPENDIX 1
Abbreviations
Ab—Antibody
Ag—Antigen
ABTS—2,2¢-Azino-bis[3-ethyl-benzothiazoline-6-sulfonic acid]diammonium salt
Anti-Ig—anti-immunoglobulin
AP—Alkaline phosphatase
BSA—Bovine serum albumin
CSPD—Disodium 3-(4-methoxyspiro{1,2-dioxetane-3,2¢-(5¢-chloro)tricyclo[3.3.1.13,7]decan}-4-yl)phenyl phosphate
EIA—Enzyme immunoassay
ELISA—Enzyme-linked immunosorbent assay
HRP—Horseradish peroxidase
Ig—Immunoglobulin
ITM—Immunological test methods
MG—4-Methylumbelliferyl galactoside
NG—Nitrophenyl galactoside
OPD—o-Phenylenediamine dihydrochloride
PBS—Phosphate-buffered saline
PVDF—Polyvinylidene fluoride
pNPP—para-Nitrophenyl phosphate
TMB—3,3¢,5,5¢-Tetramethylbenzidine
APPENDIX 2
Additional Sources of Information about Specific Topics in Validation and Data Analysis
| Analytical Data—Interpretation and Treatment |
Design and Analysis of Biological Assays |
Validation of Compendial Procedures |
Biological Assay Chapters |
|
|---|---|---|---|---|
| Means | X | |||
| Standard deviations | X | |||
| Standard errors | X | |||
| Non-normality | X | X | ||
| Nonconstant variance |
X | X | ||
| Outliers | X | X | ||
| Tests | X | |||
| Confidence intervals | X | X | ||
| Validation | X | X | ||
| Combining results from multiple assays |
X | X |
1
Capture reagent binding, blocking, analyte binding, detector antibody binding, and analysis are the five basic steps in an ELISA. Capture reagent binding, blocking, and analyte binding steps are each followed by a washing step to remove unbound reagents before the addition of the next reagent. Before analysis an appropriate substrate is added, followed by measurement of the substrate by appropriate equipment for detection. Quantitation of unknowns takes place by comparison with a standard curve.
2
The type of ELISA format depends on the availability of reagents, the intended purpose of the assay, and the physicochemical characteristics of the analyte of interest. For a Bridging ELISA, the capture and detector antibodies recognize the same epitope, and therefore the target antigen must have at least two epitopes available for binding.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Maura C Kibbey, Ph.D.
Senior Scientific Liaison, Biologics & Biotechnology (301) 230-6309 |
(GCBA2010) General Chapters - Biological Analysis |
USP38–NF33 Page 1125
Pharmacopeial Forum: Volume No. 37(5)