- British Pharmacopoeia Volume I & II
- Monographs: Medicinal and Pharmaceutical Substances
Terconazole |
 |

(Ph Eur monograph 1270)

C26H31Cl2N5O3 532.5 67915-31-5
Antifungal.
Ph Eur
1-[4-[[(2RS,4SR)-2-(2,4-Dichlorophenyl)-2-[(1H-1,2,4-triazol-1-yl)methyl]-1,3-dioxolan-4-yl]methoxy]phenyl]-4-(1-methylethyl)piperazine.
99.0 per cent to 101.0 per cent (dried substance).
White or almost white powder.
Practically insoluble in water, freely soluble in methylene chloride, soluble in acetone, sparingly soluble in ethanol (96 per cent).
It shows polymorphism (5.9).
First identification A.
Second identification B, C.
A. Infrared absorption spectrophotometry (2.2.24).
Comparison terconazole CRS.
If the spectra obtained in the solid state show differences, dissolve the substance to be examined and the reference substance separately in the minimum volume of acetone R, evaporate to dryness in a current of air and record new spectra using the residues.
B. Thin-layer chromatography (2.2.27).
Test solution Dissolve 30 mg of the substance to be examined in methanol R and dilute to 5 ml with the same solvent.
Reference solution (a) Dissolve 30 mg of terconazole CRS in methanol R and dilute to 5 ml with the same solvent.
Reference solution (b) Dissolve 30 mg of terconazole CRS and 30 mg of ketoconazole CRS in methanol R and dilute to 5 ml with the same solvent.
Plate TLC octadecylsilyl silica gel plate R.
Mobile phase ammonium acetate solution R, dioxan R, methanol R (20:40:40 V/V/V).
Application 5 µl.
Development In an unsaturated tank over a path of 10 cm.
Drying In a current of warm air for 15 min.
Detection Expose to iodine vapour until the spots appear and examine in daylight.
System suitability Reference solution (b):
- — the chromatogram shows 2 clearly separated spots.
Results The principal spot in the chromatogram obtained with the test solution is similar in position, colour and size to the principal spot in the chromatogram obtained with reference solution (a).
C. To 30 mg in a porcelain crucible add 0.3 g of anhydrous sodium carbonate R. Heat over an open flame for 10 min. Allow to cool. Take up the residue with 5 ml of dilute nitric acid R and filter. To 1 ml of the filtrate add 1 ml of water R. The solution gives reaction (a) of chlorides (2.3.1).
- 0.10° to + 0.10°.
Dissolve 1.0 g in methylene chloride R and dilute to 10 ml with the same solvent.
Liquid chromatography (2.2.29).
Test solution Dissolve 0.100 g of the substance to be examined in methanol R and dilute to 10.0 ml with the same solvent.
Reference solution (a) Dissolve 2.5 mg of terconazole CRS and 2.0 mg of ketoconazole CRS in methanol R and dilute to 100.0 ml with the same solvent.
Reference solution (b) Dilute 1.0 ml of the test solution to 100.0 ml with methanol R. Dilute 5.0 ml of this solution to 20.0 ml with methanol R.
- — size: l = 0.1 m, Ø = 4.6 mm;
- — stationary phase: base-deactivated octadecylsilyl silica gel for chromatography R (3 µm).
- — mobile phase A: 3.4 g/l solution of tetrabutylammonium hydrogen sulphate R;
- — mobile phase B: acetonitrile R1;
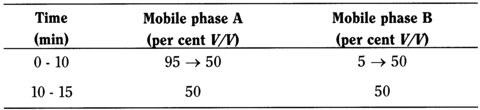
Flow rate 2 ml/min.
Detection Spectrophotometer at 220 nm.
Equilibration With acetonitrile R1 for at least 30 min and then with the mobile phase at the initial composition for at least 5 min.
Injection 10 µl; inject methanol R as a blank.
Retention time Ketoconazole = about 6 min; terconazole = about 7.5 min.
System suitability Reference solution (a):
- — resolution: minimum 13 between the peaks due to ketoconazole and terconazole; if necessary, adjust the concentration of acetonitrile in the mobile phase or adjust the time programme for the linear gradient elution.
- — impurities A, B: for each impurity, not more than the area of the principal peak in the chromatogram obtained with reference solution (b) (0.25 per cent);
- — total: not more than twice the area of the principal peak in the chromatogram obtained with reference solution (b) (0.5 per cent);
- — disregard limit: 0.2 times the area of the principal peak in the chromatogram obtained with reference solution (b) (0.05 per cent).
Maximum 0.5 per cent, determined on 1.000 g by drying in an oven at 105 °C.
Maximum 0.1 per cent, determined on 1.0 g.
Dissolve 0.150 g in 70 ml of a mixture of 1 volume of anhydrous acetic acid R and 7 volumes of methyl ethyl ketone R. Titrate with 0.1 M perchloric acid, determining the end-point potentiometrically at the 2nd point of inflexion (2.2.20).
1 ml of 0.1 M perchloric acid is equivalent to 17.75 mg of C26H31Cl2N5O3.
Protected from light.
Specified impurities A, B.
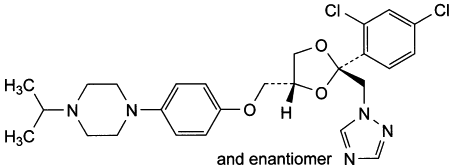
A. 1-[4-[[(2RS,4RS)2-(2,4-dichlorophenyl)-2-[(1H-1,2,4-triazol-1-yl)methyl]-1,3-dioxolan-4-yl]methoxy]phenyl]-4-(1-methylethyl)piperazine,
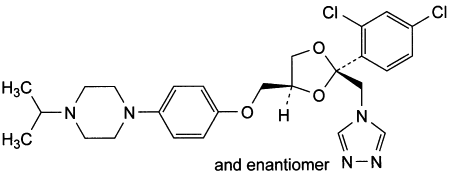
B. 1-[4-[[(2RS,4SR)-2-(2,4-dichlorophenyl)-2-[(4H-1,2,4-triazol-4-yl)methyl]-1,3-dioxolan-4-yl]methoxy]phenyl]-4-(1-methylethyl)piperazine.
Ph Eur